
Cardiac Complications in Patients with Propionic Acidemia
Moises Rodriguez-Gonzalez1*, Alvaro Antonio Perez-Reviriego2, Ana Castellano-Martinez2, Helena Maria Cascales-Poyatos2
1Pediatric Cardiology Department, Puerta del Mar Universitary Hospital, Cadiz, Spain
2Pediatrics Department, Puerta del Mar Universitary Hospital, Cadiz, Spain
Abstract
Propionic acidemia, is an autosomal recessive disorder due to the deficiency of the enzyme propionyl?coenzyme A carboxylase, which is a critical component for the metabolism of certain amino acids and lipids. The clinical complications are varied and may present at any time in the patient's life, mainly the neurological symptoms. Outside the central nerve system, haematological abnormalities including anaemia, neutropenia, thrombocytopenia or pancytopenia, immune defects, osteoporosis and pancreatitis are other rare complications reported. Of note, cardiac diseases have been recognized as increasing and life-threatening manifestations, including cardiomyopathy and electrophysiological changes such as prolongation of the QT interval.
The possible mechanisms of propionic acidemia?associated cardiac disorder, and the importance of appropriate management and early recognition, are discussed.
Introduction
Propionic acidemia (PA; OMIM #606054) is an inherited autosomal recessive inborn error of the propionate metabolism resulting from propionyl-CoA carboxylase (PCC) deficiency, which is critical for the metabolism of certain amino acids and lipids1. It was initially described in 1961 and characterised by elevations in glycine in both plasma and urine2,3.
It is a relatively rare disease with an estimated incidence of 1/100000 people, but the exact prevalence may be higher because undocumented cases of PA may cause many neonatal or fetal deaths. PCC is a dodecameric enzyme complex, consisting of 6α- and 6β-subunits, which are encoded by the PCCA and PCCB genes (mapped to chromosome 13q32 and 3q13.3-q22), respectively4,5. Mutations in either of the two genes result in PA.
PCC is a biotin-dependent enzyme located in the mitochondrial matrix involved in the metabolism of propionate, an intermediary metabolite from the breakdown of 4 essential amino acids (methionine, threonine, valine, isoleucine), odd-chain fatty acids and the side chain of cholesterol. An additional source of propionyl-CoA derives from the colonic anaerobic bacterial fermentation of carbohydrates. PCC catalyses the conversion of propionyl-CoA to methylmalonyl-CoA, which is further metabolised to succinyl-CoA, a substrate of the citric acid cycle (Figure 1).
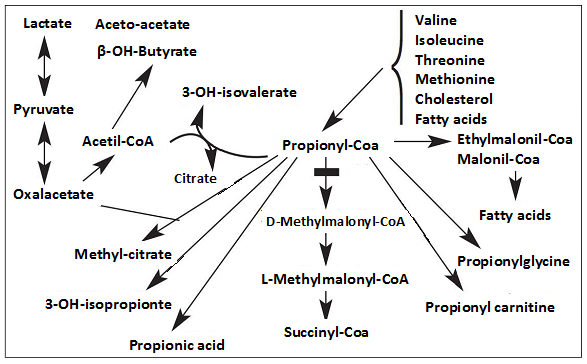
Figure 1: Precursors of propionyl-CoA and main metabolites produced66,67.
In PA, the impaired conversion of propionyl-CoA to methylmalonyl-CoA leads not only to intracellular accumulation of propionyl-CoA but also to metabolites such as 3-OH-isopropionate, glycine, propionylglycine, propionylcarnitine, fatty acids, propionic acid, and 2-methylcitrate. These metabolites have been termed organic acids, and accordingly, inherited disorders with the accumulation of these metabolites are called organic acidurias (OADs). Also, the deficiency of succinyl-CoA results in the TCA cycle and oxidative phosphorylation dysfunction, leading to an impaired mitochondrial ATP production. Therefore, end-organ injury in PA occurs due to both, the primary toxicity of the accumulating primary6,7,8,9 and secondary metabolites, and deficiency of succinyl-CoA resulting in the TCA cycle and oxidative phosphorylation dysfunction. So, PA results in multi-systemic chronic disease, mainly affecting the highly energetic organs such as the brain, muscle and heart.
Propionic acidemia is often diagnosed in the neonatal period, with characteristic patterns for organic acids and acylglycine in urine. The assessment for propionic acidemia at birth is routine in some territories as a component of newborn screening programs10,11; however, it is not a component of assessments conducted arround the world. Diagnosis is confirmed with molecular genetic testing for the PCCA and PCCB mutations.
There is a great genetic heterogeneity that produces varying degrees of functioning PCC, and therefore, the clinical picture can vary between severe early-onset forms during neonatal period or early infancy, and mild or asymptomatic late-onset variations in older children and adults. Most patients present an early onset characterize by early life-threatening decompensation including hypotonia, vomiting, ketoacidosis, hyperammonaemic coma, pancytopenia with frequent neurological sequelae including mental retardation, epilepsy and spasticity12-14; The debut occurs some days after the onset of feeding. Late-onset forms are less frequent, and they are characterised by recurrent life-threatening episodes of metabolic crises characterised for encephalopathy, lethargy, seizures, vomiting, deshidratation, metabolic acidosis, hypoglycaemia and hyperammonemia, in the context of catabolic situations, usually concurrent infections or dietetic transgressions. Patients can be asymptomatic intercrisis, but most of them suffer from various neurological deficits. Also, late-adult presentations with predominantly neurological symptoms and asymptomatic individuals have been described12-14.
PA is a multi-system disease, so complications involve many different organs and organ systems, but mainly the central nervous system. Thus, encephalomyopathy is typically the most prominent manifestation. Children with PA may have a failure to thrive, muscular hypotonia, developmental delay, psychomotor retardation, retardation in speech development, and epilepsy15. Also, they usually develop stroke-like episodes of the basal ganglia infarction of basal ganglia in acute metabolic crises, leading to abnormal movements such as ataxia, dystonia or choreoathetosis. Less common, visual impairment secondary to optic atrophy, hearing loss, and development of comorbidities such as attention-deficit disorder, autism, anxiety or acute psychosis, have been reported. Outside the central nervous system, haematological abnormalities including anaemia, neutropenia, thrombocytopenia or pancytopenia16,17, immune defects18, osteoporosis19,20 and pancreatitis are other rare complications reported. Of note, cardiac diseases have been recognised as an increasing and vital manifestation of PA21,22.
Cardiac Complications in PA
Cardiac complications in patients with PA are well recognised and include mainly cardiomyopathy and acquired long QT syndrome (LQTS).
Cardiomyopathy
Dilated cardiomyopathy (DCM) is a cardiac condition characterised by dilatation of the left ventricle or both ventricles and systolic dysfunction. DCM has an estimated prevalence of 1:250 and is one of the leading causes of heart failure that requires heart transplantation23. In children, DCM comprises some incompletely defined aetiologies, including congenital heart defects, cardiac inflammation with or without infectious agents, cytotoxic medication or drugs, arrhythmias and genetic causes. DCM is diagnosed in the presence of left ventricular diastolic diameter > 2 Z score for body surface area of the patient in the echocardiography, and may be or not associated with systolic dysfunction and heart failure24.
Of note, cardiomyopathy as a complication of organic acidemias, and particularly of propionic acidemia, is now a well-accepted phenomenon25. It has been recognised as a complication of PA since 1993, when Massoud et al. reported 19 patients with PA of whom, 6 developed dilated cardiomyopathy, with remarkably high mortality of 50%26. Later, approximately 30 cases of DCM complicating 9-37% of PA cases, with a mortality of 40-50%, have been reported, so this complication is not universal in the natural history of PA. Also, no specific time-course has been observed between disease presentation and development of cardiomyopathy, with nearly 50% of cases affecting patients with early onset and nearly 50% with late onset, and mean age at diagnosis of 6.5 years, ranging from 3 weeks to 25 years. In addition, cardiomyopathy appears to be unrelated to clinical features or metabolic markers, and remarkably, it is an on-going and irreversible proccess. Although rare, isolated cardiac involvement as the unique manifestation of PA has been reported in teenagers and adults with DCM who underwent metabolic screening. Even some cases received cardiac transplantation before to be diagnosed with PA. Most cases (>90%) are dilated cardiomyopathy, but hypertrophic and non-compacted cardiomyopathy cases have also been reported27.
Long QT syndrome (LQTS)
Long QT syndrome is a rare condition that can be caused by a genetic abnormality or acquired reasons. Of note, it predisposes affected individuals to prolongation of ventricular repolarization, which can result in fatal ventricular arrhythmia (Torsade de Pointes is its hallmark arrhythmia) leading to syncope, near syncope, palpitations, seizures or sudden death, the later even as the initial manifestation. Congenital LQTS is a channelopathy genetically determined with a population prevalence of 1/2000 that. This condition may be inherited in an autosomal dominant (Romano-Ward syndrome) or an autosomal recessive (Jervell and Lange-Nielsen syndrome) form11. Many subtypes are the result of mutations in ion channel subunits or regulatory protein-coding genes. LQT1, LQT2 and LQT3 genotypes with mutations involving KCNQ1, KCNH2 and SCN5A respectively, comprise more than 90% of genotype-positive cases of LQTS28. The responsible genes are located on chromosomes 11, 7 and 3p-21, respectively. Other types of LQTS with significantly less prevalence also exist. The genetic background of 25% of patients with LQTS remains elusive. LQTS is diagnosed in the presence of either: 1. LQTS risk score (Schwartz) of at least 3.5, in the absence of a secondary cause for QT prolongation (medications, electrolyte disturbance, increased thyroid hormone concentrations, left ventricular hypertrophy, ischemia, and slow heart rate). 2. Unequivocally pathogenic mutation in one of the LQTS genes. 3. QTc at least 500 ms on repeated 12-lead ECGs in the absence of a secondary cause for QT interval prolongation. 4. QTc between 480 and 499 ms on repeated 12-lead ECGs in a patient with unexplained syncope in the absence of a secondary cause for QT interval prolongation and the absence of a pathogenic mutation.
Acquired long QT syndrome as a complication of PA is also known since 1993, when Massoud et al. reported the first case, in a series of 19 PA patients26. After this report, approximately 16 cases of acquired LQTS associated with PA have been reported. In 2006 Kakavand described one asymptomatic 7-year-old patient who was incidentally diagnosed with LQTS when she was studied to discard cardiomyopathy29. Baumgather et al. presented in 2007 a longitudinal observational study of 10 patients with PA under cardiology follow-up, and demonstrated that 70% of children after infancy had prolonged QTc > 440 ms and 60% had a noticeably prolonged QTc > 460 ms30. Preterminal T-wave inversion, which was related with a QTc > 460 ms was present in 60% of patients. Also, 20% had rhythm problems such as ventricular ectopic beat. No life-threatening arrhythmias were reported. Of note, Jameson et al. in 200831, published the first and the only case report of a child with PA and long-QT syndrome suffering a life-threatening event secondary to long QT syndrome. In 2013 grunter et al. retrospectively reviewed 55 cases of PA and found that LQTS was present in only 22% of cases, with no cases of life-threatening arrhythmias32. However, ECG register was not obtained in all patients, and no exercise and 24h ECGs were performed. Afterwards, we published a 9-year-old boy who complained of complained of palpitations and near-syncope episodes related with exercise33. A prolonged QTc interval was noted on ECG-Holter. Although no life-threatening arrhythmias were detected, we think that ventricular arrhythmias could be the responsibility of the symptoms of the patient. Recently, Duras et al. et al. described two sisters with PA who have prolonged QT duration that was incidentally detected in an outpatient setting34.
Pathophysiologic Mechanisms of Cardiac Complications in PA
Although long-term cardiac complications are well-recognised with an increasing number of reported cases in PA patients during recent years, the pathophysiological mechanisms underlying the PA-associated cardiomyopathy and LQTS remains unclear. In this context, several possible mechanisms have been discussed, and cardiac complications might be the result of multifactorial processes35.
Carnitine or biotin deficiency
Deficiency in free carnitine, as well as biotin deficiency, have been suggested as potential risk factors for the development of cardiomyopathy and arrhythmias in PA patients. However, it seems to be unlikely, because with the advent of carnitine and biotin therapy36,37, plasma levels are usually maintained in the normal range, and despite this cardiac disturbances persist, what reduces the probability of carnitine or biotin deficiency as the basis of the problem. Of note, Massoud et al. documented total and free carnitine concentrations to be low in heart muscle despite carnitine supplementation and repeatedly normal plasma carnitine levels26. So, the matter seems to be an impaired carnitine uptake into the heart, but the exact responsible mechanism is not clear26. Therefore, carnitine or biotin deficiency has not been a prime suspect of cardiac dysfunction and arrhythmias in PA.
Genetics
Recently the possibility that some patients with PA have a genetic abnormality that is a variant of the congenital LQTS has been proposed. However, QTc interval prolongation is not permanently present in patients with PA38,25, and the younger patients seem less affected than, the older ones, suggesting that cardiac complications are an ongoing progressive and not a congenital process.
Long-term and sustained mitochondrial dysfunction
Impairment of energy metabolism, secondary to chronic mitochondrial dysfunction due to mechanisms such as toxic metabolites direct damage, deficiency of essential metabolic intermediates, and to concomitantly increased respiratory oxidative stress, is thought to play a crucial role in the development of both cardiomyopathy and LQTS in patients with PA (Figure 2).
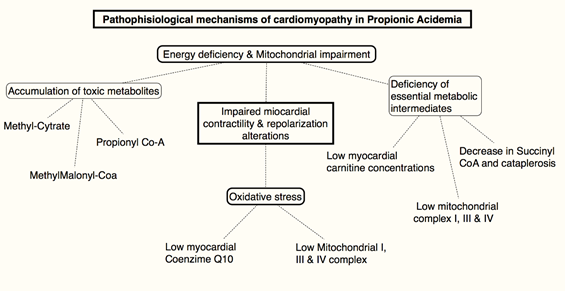
Figure 2: Pathophisiological mechanisms of cardiomyopathy and long QT in PA
Energetic deficiency
The normal mitochondrial function requires sufficient energy production through the Tricarboxilic acid cycle (TCA) and oxidative phosphorylation. Therefore, PA result in multi-systemic chronic disease, particularly in the highly energetic organs such as brain, heart, kidney, and eye. End-organ injury occurs due to both primary toxicity of both the accumulating primary and secondary metabolites and deficiency of succinyl-CoA (a key chemical in the TCA cycle) resulting in TCA cycle and oxidative phosphorylation dysfunction.
The heart function is thought to rely on propionate as an anaplerotic metabolite mainly. Via this anaplerotic pathway, succinyl-CoA is fuelled into the TCA cycle, leading to adenosine triphosphatase (ATP) production (Figure 3).
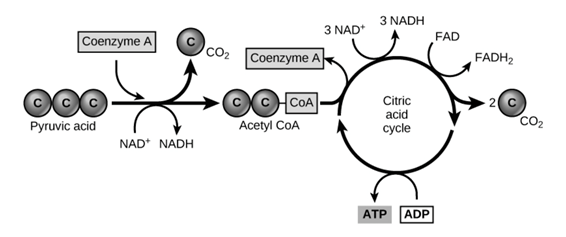
Figure 3: Schematic representation of TCA cycle (also named citric acid cycle).
The primary enzymatic defect in patients with PA is located in the final steps of propionate metabolism. So, the inherited deficiency of the anaplerotic propionate metabolism could lead to cardiac manifestations of PA. Of note, similar problem occurs in patients with genetic disorders of long chain fatty acid oxidation. These patients also develop a deplection of the TCA cyclce substrate due to a secondary deficiency of propionate (Figure), that results in symptoms that include cardiomyopathy and arrythmia. The usual treatment of long chain FAOs is with medium chain triglyceride oil, which can bypass the metabolic block, but doesn’t address the propionate deficiency. Recently, several studies have reported positive clinical results following anaplerotic therapy with triheptanoin in this patients. Triheptanoin is a triglyceride composed of three seven-carbon fatty acids, heptanoate, esterified to a glycerol backbone. Oxidation of one heptanoate yields two acetyl-CoA molecules and one of propionyl-CoA. Propionyl-CoA is converted to succinyl-CoA, which is an intermediate of the TCA cycle and subsequently increases the intermediate pool size (figure 4).
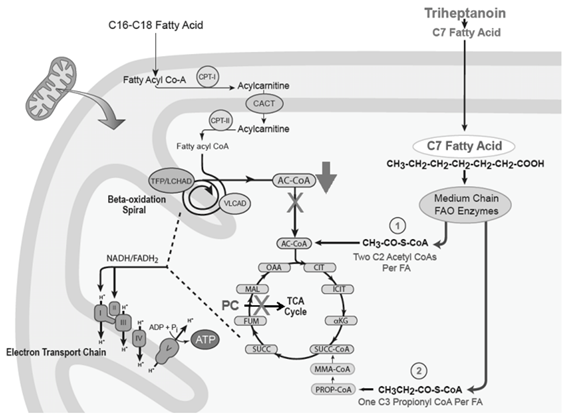
Figure 4: Schematic representation of TCA cycle deplection in long chain fatty acid oxidation defects, along with its reversal by the anaplerotic agent triheptanoin.
The biological effect of the inherited deficiency of the anaplerotic propionate, metabolism is aggravated by synergistic secondary effects of accumulating toxic metabolites on energy metabolism39. Deficiency of pyruvate dehydrogenase complex (PDHc), citrate synthase (CS), aconitase, 2- oxoglutarate dehydrogenase complex (OGDHc), succinate-CoA ligase (ligase), and the mitochondrial succinate transport due to inhibition by accumulating propionyl-CoA (P-CoA) and 2-methylcitrate (2-MCA), result in a synergistic impairment of energy metabolism, with a particular focus on the TCA cycle. Increased intracellular propionyl-CoA ester and its metabolites levels in heart, raise a possible direct acute toxicity of propionylcarnitine leading to a cataplerotic state and a secondary impairment of energy metabolism40. Other metabolites, including 3-hydroxypropionate, methylcitrate and glycines, may also exert cardiotoxicity at higher concentrations, although this has not been elucidated to date. Moreover, toxic metabolites, inhibiting the pyruvate dehydrogenase and the succinyl-CoA ligase, may compromise the TCA cycle, and interfere with ureagenesis inducing hyperammonemia via inhibition of N-acetylglutamate synthase41.
Increased oxidative stress
The inhibition of the oxidative phosphorylation in mitochondria by propionyl-CoA, could lead to increased reactive oxygen species which may impair mitochondrial DNA expression42,43. Thus, multiple deficiencies of respiratory chain complexes were found in cardiac cells of patients with PA44,45. Reported mitochondrial abnormalities in PA supporting this mechanism include low myocardial free carnitine, and low activity of myocardial complex complex III, complex IV, providing evidence of functional impairment46. Also low myocardial CoQ10 in cardiomyopathy in PA, highlighting secondary mitochondrial impairment as a relevant causative mechanism47. This notion is supported by the finding of multiple oxidative phosphorilation deficiencies, in particular, deficiency of bc1 complex and cytochrome c oxidase in the liver, skeletal muscle, heart muscle, and kidney of patients with PA48. Also, decreased antioxidative defence due to glutathione depletion, and epigenetic modifications due to propionyl-CoA-induced histone acetylation and thus permanent alteration of gene expression are likely candidate mechanisms. Taken together, all of these findings strongly support secondary respiratory chain impairment in the myocardium as one of the main pathophysiological explanations for PA-associated cardiomyopathy and LQTS.
Management of PA Patients with Cardiac Complications
Treatment strategies for PA patients are reflective of their clinical state, aimed at addressing the disease specific complications of the initial acute presentation, long-term management, and intermittent metabolic decompensations that can occur from various triggers.
The management of PA and its cardiac complications is largely supportive. During acute illness (metabolic crises), the mainstay of treatment is to reverse the catabolic state and prevent accumulation of propionic compounds, which involves treating the underlying illness, providing calories via intravenous glucose and lipids49,50, and cessation of protein intake for up to 48 hours51,52.
Long-term treatment principally involves adherence to a protein-restricted diet, as well as supplementation with a “mitochondrial cocktail” that would include coenzyme Q10 (CoQ10), creatine, L-carnitine, thiamine, riboflavin, folate, and other antioxidants such as vitamins C and E. Metronidazole53,54, which reduces intestinal flora that produce propionic acid through fermentation of carbohydrates, also have been used in the treatment for PA.
The management of cardiac complications, including heart failure, bradyarrhythmias, and tachyarrhythmias, follows the same guidelines as those for the general population. Therapy with β-blockers and angiotensin-converting enzyme inhibitors should be considered in all patients who have cardiomyopathy at the time of discharge from the hospital, even though there are no randomised controlled trials to support the use of such therapies in this population. Diuretics are prescribed for alleviating congestive symptoms. In selected patients who have advanced heart failure due to cardiomyopathy, cardiac transplantation can be considered55-59. Three pediatric patients with mitochondrial cardiomyopathy who underwent cardiac transplantation reportedly had excellent early and late outcomes24,60,61.
The mainstay of therapy in LQTS is the prophylactic, regular and uninterrupted use of beta-blockers34,62,63. The international LQTS registry has produced a series of observational research studies demonstrating a reduced risk among those who take beta-blockers. This treatment is recommended for patients diagnosed as LQTS who are asymptomatic with QTc ≥470 ms and symptomatic for syncope or documented ventricular tachycardia/ventricular fibrillation34,62,63. Propranolol (2–4 mg/kg/day) has been shown to be effective in suppressing recurrent cardiac events. The most significant benefit occurs mainly in those who have had recent syncope or cardiac arrest before age 7 or a very prolonged QT interval (> 500 ms). Patients with a history of aborted cardiac arrest, symptomatic patients in the first year of life and patients with J-LNS carrying KCNQ1 mutations, are at unusually high risk and therefore, implantable cardioverter defibrillator (ICD) and left cardiac sympathetic denervation (LCSD) might be necessary8. Besides medical treatment, lifestyle advice and avoidance of QT-prolonging drugs (www.qtdrugs.org or http://www.crediblemeds.org) are essential63.
Finally, the use of liver transplantation (LT) for patients with PA has been described in several case study series as one of the potential treatment modalities to restore some enzyme activity, because liver is the major site of branched chain amino acid metabolism and subsequent propionic acid production63-68. Given that the enzymes responsible for PA, it was not expected that LT would provide a metabolic cure; rather, LT was proposed as a way to stabilize metabolically fragile patients, with early onset and repeated decompensations, minimizing the risk of further decompensations, and improving quality of life (no need of full dietary restrictions)63-68. LT in patients with PA may be considered a valid and long-lasting treatment when cardiomyopathy is progressive and unresponsive to medical therapy. Currently available data from our experience and literature are limited but suggest that liver transplantation may have a positive and lasting effect on PA-related cardiomyopathy, otherwise life-threatening. However, robust data on transplant experiences with these rare diseases remains sparse and with controversial results. Of note, althought it has been previously shown that the PA-associated cardiomyopathy is reversible after LT, early and severe complications difficulties in perioperative period (heart failure, adult respiratory distress síndrome, renal failure, hepatic artery thrombosis) and a high mortality (58%) with a graft survival rate of 60% at 5 year, have been reported68.
Conclusion
PA, cardiomyopathy and LQTS, are rare disorders that are potentially lethal separately so, this association increases the risk of life-threatening cardiac events and mortality in these patients. Their development is independent of the metabolic control, and the pathomechanisms are not yet well understood, but an impaired mitochondrial function in the myocardium through different pathways seems to be one of the main pathophysiological explanations. Regular electrocardiographic and echocardiographic investigations in all these patients are warranted, including at least yearly ECGs with a determination of QTc and regular 24-hour Holter monitoring to detect life-threatening, but treatable ventricular arrhythmias and cardiac dysfunction. When LQTS or CMP are diagnosed, therapy follows the same guidelines as those for the general population. The evidence is increasing that adult patients with PA may develop late-onset organ manifestation even if they have been considered as metabolically stable for years. These clinical observations emphasise the need for establishing new therapeutic concepts and for revaluating available strategies to protect against these long-term complications. Liver transplantation is a promising therapy that can reverse the cardiac complications of PA, but evidence regarding safety and efficacy is still scarce.
Contributors' Statement: Drs. Rodriguez-Gonzalez conceptualized and designed the study, drafted the initial manuscript, and approved the final manuscript as submitted. Drs. Castellano-Martinez, Perez-Reviriego and Cascales-Poyatos, reviewed and revised the manuscript, and approved the final manuscript as submitted. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
References
- Deodato F, Boenzi S, Santorelli FM, et al. Methylmalonic and propionic aciduria. Am J Med Genet C Semin Med Genet. 2006 May 15; 142C(2): 104-12.
- Childs B, Nyhan WL, Borden M, et al. Idiopathic hyperglycinemia and hyperglycinuria: a new disorder of amino acid metabolism I. Pediatrics. 1961; 27: 522–538.
- Nyhan WL, Borden M, Childs B. Idiopathic hyperglycinemia: a new disorder of amino acid metabolism. II. The concentrations of other amino acids in the plasma and their modification by the administration of leucine. Pediatrics. 1961; 27: 539–550.
- Ugarte M, Pérez-Cerdá C, Rodríguez-Pombo P, et al. Overview of mutations in the PCCA and PCCB genes causing propionic acidemia. Hum Mutat. 1999; 14: 275.
- Kraus JP, Spector E, Venezia S, et al. Mutation analysis in 54 propionic acidemia patients. J Inherit Metab Dis. 2012; 35: 51.
- Schwab MA, Sauer SW, Okun JG, et al. Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem J. 2006; 398: 107–112.
- Cheema-Dhadli S, Leznoff CC, Halperin ML. Effect of 2-methylcitrate on citrate metabolism: implications for the management of patients with propionic acidemia and methylmalonic aciduria. Pediatr Res. 9 (1975) 905–908.
- M. Brock, W. Buckel, On the mechanism of action of the antifungal agent propionate, Eur. J. Biochem. 2004; 271: 3227–3241.
- Rutledge SL, Geraghty M, Mroczek E, et al. Tubulointerstitial nephritis in methylmalonic acidemia. Pediatr Nephrol. 1993; 7: 81–82.
- Pérez-Cerdá C, Pérez B, Merinero B, et al. Prenatal diagnosis of propionic acidemia. Prenatal Diagnosis. Wiley-Blackwell. 2004 Dec 21; 24(12): 962–4.
- Buchanan PD, Kahler SG, Sweetman L, et al. Pitfalls in the prenatal diagnosis of propionic acidemia. Clin Genet. 1980 Sep; 18(3): 177-83.
- Nyhan WL, Bay C, Beyer EW, et al. Neurologic nonmetabolic presentation of propionic acidemia. Arch Neurol. 1999; 56: 1143–1147. [17]
- Chemelli AP, Schocke M, Sperl W, et al. Magnetic resonance spectroscopy (MRS) in five patie.
- Haberlandt E, Canestrini C, Brunner-Krainz M, et al. Epilepsy in patients with propionic acidemia. Neuropediatrics. 2009; 40: 120–125.
- Wolff B, Hsia YE, Sweetman L, et al. Propionic acidemia: a clinical update. J Pediatr. 1981; 99: 835–846.
- Stork LC, Ambruso DR, Wallner SF, et al. Pancytopenia in propionic acidemia: hematologic evaluation and studies of hematopoiesis in vitro. Pediatr Res. 1986; 20: 783–788.
- Sweetman L, Nyhan WL, Cravens J, et al. Propionic acidaemia presenting with pancytopaenia in infancy. J Inherit Metab Dis. 1980; 2: 65–69.
- Sipahi T, Yilmaz D, Tavil B. Propionic acidemia with myelodysplasia and neutropenia in a Turkish child. J Pediatr Hematol Oncol. 2004; 26: 154–155.
- Ozand PT. Hypoglycemia in association with various organic and amino acid disorders. Semin Perinatol. 2000; 24: 172.
- Talbot JC, Gummerson NW, Kluge W, et al. Osteoporotic femoral fracture in a child with propionic acidaemia presenting as non-accidental injury. Eur J Pediatr. 2006; 165: 496.
- Mardach R, Verity MA, Cederbaum SD. Clinical, pathological, and biochemical studies in a patient with propionic acidemia and fatal cardiomyopathy. Mol Genet Metab. 2005; 85: 286.
- Pena L, Burton BK. Survey of health status and complications among propionic acidemia patients. Am J Med Genet A. 2012; 158A: 1641.
- Ameloot K, Vlasselaers D, Dupont M, et al. Left ventricular assist device as bridge to liver transplantation in a patient with propionic acidemia and cardiogenic shock. J Pediatr. 2011 May; 158(5): 866–7–authorreply867.
- Laemmle A, Balmer C, Doell C, et al. Propionic acidemia in a previously healthy adolescent with acute onset of dilated cardiomyopathy. Eur J Pediatr. 8 ed. 2014 Jun 11; 173(7): 971–4.
- Bhan AK, Brody C. Propionic acidemia: a rare cause of cardiomyopathy. Congest Heart Fail. 2001 Jul; 7(4) :218–9.
- Massoud AF, Leonard JV. Cardiomyopathy in propionic acidaemia Medical Unit, Institute of Child Health, London, WC1N 1EH. United Kingdom Eur J Pediatr. 1993; 152: 441-445.
- Lee TM, Addonizio LJ, Barshop BA, et al. Unusual presentation of propionic acidaemia as isolated cardiomyopathy. J Inherit Metab Dis. 8 ed. 2009 Feb 24; 32(S1): 97–101.
- David J Tester BS, Michael J Ackerman MD PhD. GENETICS OF LONG QT SYNDROME Methodist Debakey Cardiovasc J. 2014 Jan-Mar; 10(1): 29–33.
- Kakavand B, Schroeder VA, Di Sessa TG. Coincidence of Long QT Syndrome and Propionic Acidemia Pediatr Cardiol. Springer New York. 2006 Feb 1; 27(1): 160–1.
- Baumgartner D, Scholl-Bürgi S, Sass JO, et al. Prolonged QTc intervals and decreased left ventricular contractility in patients with propionic acidemia. J Pediatr. 2007; 150: 192–197.
- Jameson E, Walter J. Cardiac Arrest Secondary to Long QTC in a Child with Propionic Acidemia. Pediatr Cardiol. Springer-Verlag; 2007 Dec 5; 29(5): 969–70.
- Grünert SC, Müllerleile S, De Silva L, et al. Propionic acidemia: clinical course and outcome in 55 pediatric and adolescent patients. Orphanet Journal of Rare Diseases. BioMed Central. 2013 Jan 10; 8(1): 6.
- Rodriguez-Gonzalez M, Castellano-Martinez A. Long QTc Syndrome and Propionic Acidemia. Indian Pediatr. 2016 Sep 8; 53(9): 841.
- Duras E, ?rdem A, Özkaya O. Long QT syndrome diagnosed in two sisters with propionic acidemia: a case report. Journal of Pediatric Endocrinology and Metabolism. 2017 Aug 14; 30(10): 169–4.
- Sass JO, Hofmann M, Skladal D, et al. Propionic acidemia revisited: a workshop report. Clin Pediatr (Phila). 2004; 43: 837–843.
- Nyhan WL, Ozand PT. Atlas of Metabolic Diseases, 1st ed, Chapman and Hall Medical, London 1998.
- Hillman RE, Keating JP, Williams JC. Biotin-responsive propionic acidemia presenting as the rumination syndrome. J Pediatr. 1978; 92: 439.
- Bodi I, Grünert SC, Becker N, et al. Mechanisms of acquired long QT syndrome in patients with propionic Acidemia. Heart Rhythm Elsevier. 2016 Mar 24; 1–11.
- Morath MA, Okun JG, Müller IB, et al. Neurodegeneration and chronic renal failure in methylmalonic aciduria-a pathophysiological approach. J Inherit Metab Dis. 2008; 31(1): 35-43.
- Kölker S, Burgard P, Sauer SW, et al. Current concepts in organic acidurias: understanding intra- and extracerebral disease manifestation. J Inherit Metab Dis. 2013; 36: 635.
- Gebhardt B, Dittrich S, Parbel S, et al. N-carbamylglutamate protects patients with decompensated propionic aciduria from hyperammonaemia. J Inherit Metab Dis. 2005; 28: 241.
- de Keyzer Y, Valayannopoulos V, Benoist JF, et al. Multiple OXPHOS deficiency in the liver, kidney, heart, and skeletal muscle of patients with methylmalonic aciduria and propionic aciduria. Pediatr Res. 2009; 66: 91–95.
- Fragaki K, Cano A, Benoist JF, et al. Fatal heart failure associated with CoQ10 and multiple OXPHOS deficiency in a child with propionic acidemia. Mitochondrion. 2011; 11: 533–536. [60] P.J. Mc Guire, A. Parikh, G.A. Diaz, Profiling of oxidative stress in patien
- Kovacevic A, Kölker S, Hoffmann GF, et al. Cardiac Manifestation in Propionic Acidemia: A 30-Year Single-Center Experience. Georg Thieme Verlag KG. 2018; DGPK–V165.
- Fraser JL, Venditti CP. Methylmalonic and propionic acidemias: clinical management update. Current Opinion in Pediatrics. 2016 Dec; 28(6): 682–93.
- de Keyzer Y, Valayannopoulos V, Benoist JF, et al. Multiple OXPHOS deficiency in the liver, kidney, heart, and skeletal muscle of patients with methylmalonic aciduria and propionic aciduria. Pediatr Res. Nature Publishing Group. 2009 Jul; 66(1): 91–5.
- Baruteau J, Hargreaves I, Krywawych S, et al. Successful reversal of propionic acidaemia associated cardiomyopathy: Evidence for low myocardial coenzyme Q10 status and secondary mitochondrial dysfunction as an underlying pathophysiological mechanism. MITOCH Elsevier BV. 2014 Jul 1; 17(C): 150–6.
- Haines R, Crichton S, Wilson J, et al. Cardiac biomarkers are associated with maximum stage of acute kidney injury in critically ill patients: a prospective analysis. Crit Care Critical Care. 2017 Apr 4; 21(1): 1–8.
- Ekvall SW, Ekvall VK, editors. Pediatric and Adult Nutrition in Chronic Diseases, Developmental Disabilities, and Hereditary Metabolic Disorders. Oxford University Press.
- Frazier D. Methylmalonic Acidemia and Propionic Acidemia. In: Ekvall SW, Ekvall VK, editors. Pediatric and Adult Nutrition in Chronic Diseases, Developmental Disabilities, and Hereditary Metabolic Disorders. Oxford University Press; pp. 300–4. (Prevention, Assessment, and Treatment).
- van Calcar S. Nutrition Management of Propionic Acidemia and Methylmalonic Acidemia. In: Nutrition Management of Inherited Metabolic Diseases. Cham: Springer International Publishing. 2015; 221–8.
- Baumgartner MR, rster FH, Dionisi-Vici C, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. 2014 Sep 26; 1–36.
- Thompson GN, Chalmers RA, Walter JH, et al. The use of metronidazole in management of methylmalonic and propionic acidaemias. Eur J Pediatr. 1990; 149: 792–796.
- Clausen MR, Bonnen H, Tvede M, et al. Colonic fermentation to short-chain fatty acids is decreased in antibiotic-associated diarrhea. Gastroenterology. 1991; 101: 1497–1504.
- Sutton VR, Chapman KA, Gropman AL, et al. Chronic management and health supervision of individuals with propionic acidemia. Mol Genet Metab. 2012; 105: 26.
- Vara R, Turner C, Mundy H, et al. Liver transplantation for propionic acidemia in children. Liver Transpl. 2011; 17: 661.
- Saudubray JM, Touati G, Delonlay P, et al. Liver transplantation in propionic acidaemia. Eur J Pediatr. 1999; 158 Suppl 2: S65.
- Kasahara M, Sakamoto S, Kanazawa H, et al. Living-donor liver transplantation for propionic acidemia. Pediatr Transplant. 2012; 16: 230.
- Squires RH, Ng V, Romero R, et al. Evaluation of the pediatric patient for liver transplantation: 2014 practice guideline by the American Association for the Study of Liver Diseases, American Society of Transplantation and the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. Hepatology. 2014; 60: 362.
- Romano S, Valayannopoulos V, Touati G, et al. Cardiomyopathies in propionic aciduria are reversible after liver transplantation. J Pediatr. 2010; 156: 128.
- Vermeer N, Meurisse N, Vlasselaers D, et al. Liver transplantation in a patient with an intraabdominally located left ventricular assist device: surgical aspects--case report. Transplant Proc. 2012; 44: 2885.
- Riemersma M, Hazebroek MR, Enden den ATJMH-V, et al. Propionic acidemia as a cause of adult-onset dilated cardiomyopathy. Nature Publishing Group. Nature Publishing Group. 2017 Aug 28; 1–7.
- Arrizza C, De Gottardi A, Foglia E, et al. Reversal of cardiomyopathy in propionic acidemia after liver transplantation: a 10-year follow-up. Transpl Int. 2015 Sep 11; 28(12): 1447–50.
- Davison JE, Davies NP, Wilson M, et al. MR spectroscopy-based brain metabolite profiling in propionic acidaemia: metabolic changes in the basal ganglia during acute decompensation and effect of liver transplantation. Orphanet J Rare Dis. 2011 May 9; 6: 19. doi: 10.1186/1750-1172-6-19.
- Meyburg J, Hoffmann GF. Liver transplantation for inborn errors of metabolism. Transplantation. 2005 Sep 27; 80(1 Suppl): S135-7.
- Fenton WA, Rosenberg LE. Disorders of propionate and methylmalonate metabolism. Scriver, CR, Beaudet, AL, Sly, WS, y Valle, D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill New York. 2001; 8th ed: 2165-2194.
- Sanjurjo P, Aldámiz K, Prieto JA, et al. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Ed Ergon, Madrid. 2006; 377-392.
- Charbit-Henrion F, Lacaille F, McKiernan P, et al. Early and Late Complications After Liver Transplantation for Propionic Acidemia in Children: A Two Centers Study. American Journal of Transplantation. 2015 Feb 12; 15(3): 786–91.