
Chromatin remodelers, their implication in cancer and therapeutic potential
Laura Manelyte
Biochemistry Center Regensburg, Laboratory of Chromatin Dynamics and Nuclear Architecture, University of Regensburg, Universitätstraße 31, Regensburg DE-93053, Germany
Abstract
The chromatin remodeling complexes alter chromatin structures. They remodel nucleosomes in ATP-dependent manner and have essential roles in DNA damage repair, recombination, replication and transcriptional control. Increasing evidences indicate that subunits of chromatin remodelers are mutated and/or deregulated in a number of human cancers, and how they influence the cancer gene expression program during cancer initiation and progression is becomming clearer. Therefore, chromatin remodeling complexes arose as promising new targets for the treatment of human cancers. In this review, chromatin remodeling complexes, their epigenetic reader domains and available inhibitors are described. The insights into the misregulated chromatin remodelers pathways in human malignancies and the novel approach targeting deregulated chromatin remodelers to improve chemotherapy efficiency are discussed.
Chromatin remodelers, epigenetic reader domains and their therapeutic potential
A nucleosome is a basic unit of DNA packaging in humans, consisting of 146 bp DNA wrapped around a histone octamer. To control the accessibility of nucleosomal DNA target sites cells have evolved molecular machines, chromatin remodelers. Chromatin remodelers use the ATP-hydrolysis energy to alter the interactions between the nucleosomes and DNA: reposition nucleosomes, evict or influence their assembly and composition on the DNA strand and thereby regulate the cellular processes such as transcription, replication, recombination and DNA repair. Human cells carry different chromatin remodelers and to ensure the healthy status of the cell, chromatin remodelers must be in the right place, at the right time and with the right activity1.
Chromatin remodelers are composed of the catalytic subunit, which is usually accompanied by one or several accessory subunits. Catalytic subunit contains conserved ATPase domain which belong to Snf2 family of helicase-like proteins and unique protein domains2. Based on unique protein domains, chromatin remodelers fall into 4 families: SWI/SNF, ISWI, CHD and INO80 (Fig. 1)3. These families differ in their epigenetic reader domains, which recognize and bind to specifically modified histone tails. For example, SWI/SNF family remodelers contain bromodomain at their C-terminus, which binds acetylated histone tails. CHD family remodelers at their N-terminus harbor chromodomains that bind methylated histone tails. Furthermore, the complexity and specificity of chromatin remodeling complexes is achieved by their accessory subunits, that contain additional epigenetic reader domains, RNA binding domains or even form complexes with histone deacethylases (HDAC) and histone methyltransferases (HMT)2.
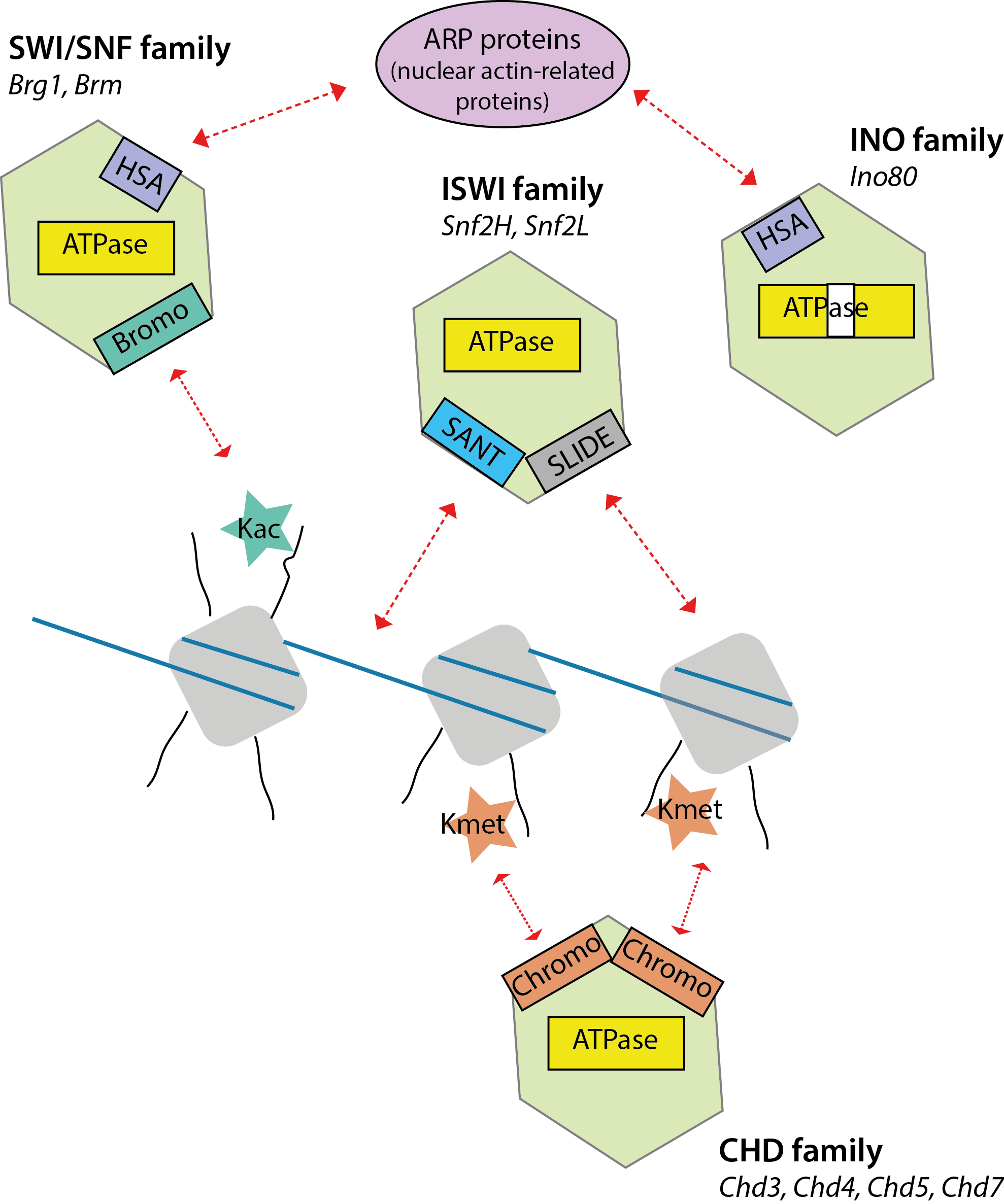
Figure 1: Catalytic subunits of chromatin remodelers fall into four families.They share conserved ATPase domain and differ in their unique protein domains. Catalytic subunit is shown in green, ATPase domain is in yellow, DNA is blue and nucleosomes are depicted in grey.
Malignant cell abuses chromatin remodelers in different ways
To meet the demands of growth, cancer cells reprogram their metabolism. The connection between cancer and chromatin remodelers arose two decades ago, when SNF5 gene, which encodes a member of the chromatin-remodelling SWI/SNF complex, was found to be mutated in malignant rhabdoid tumours4. Since then, evidence has been accumulating how the deregulated chromatin remodelers can support the uncontrolled growth of the cell.
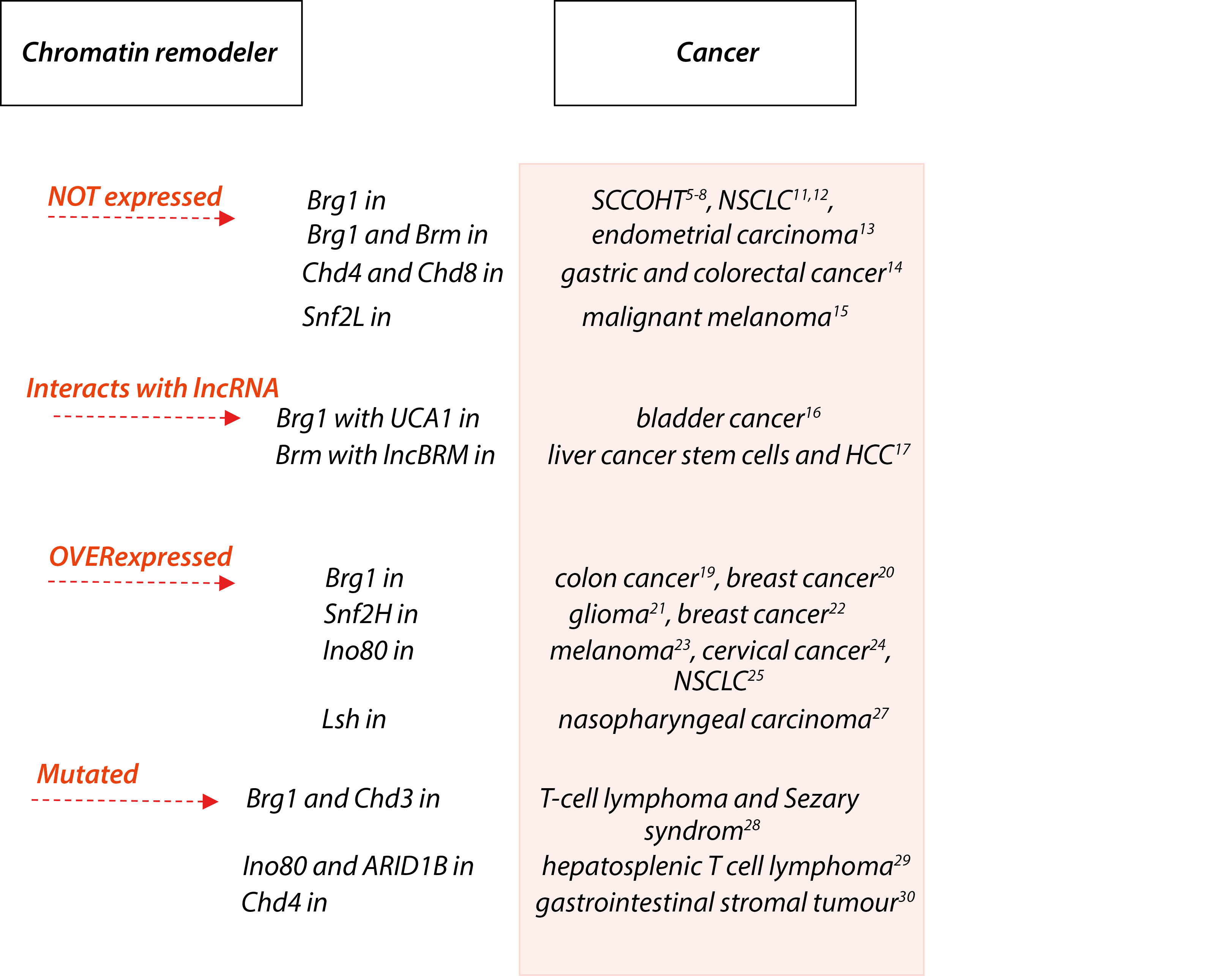
Figure 2: Malignant cells abuses chromatin remodelers in different ways. Some recent examples of deregulated catalytic subunits of chromatin remodelers.
Loss of expression: Absence of functional chromatin remodeling complexes seem to fuel tumour progression. For example, up to 90% small cell carcinoma of the ovary, hypercalcaemic type (SCCOHT) harbour inactivating mutations in the chromatin remodelling gene brg1 with concomitant loss of its encoded protein Brg1, one of two mutually exclusive ATPases of the SWI/SNF chromatin remodelling complex5–8.
Moreover, recent studies have shown that Brm, the other mutually exclusive ATPase of the SWI/SNF complex, is lost in SCCOHT as well and that restoration of either SWI/SNF ATPases can inhibit the growth of SCCOHT cell lines9,10. Brg1 was also lost or mutated in a significant number of human lung tumor cell lines and primary non-small cell lung carcinoma clinical specimens11,12. Almost one-third of endometrial undifferentiated carcinomas show loss of Brg1 and Brm expression. The majority of the Brg1-deficient cases show concominant loss of Brm expression13. Frameshift mutation and loss of expression of chd genes are common in gastric cancers (GC) and colorectal cancers (CRC) with high microsatellite instability. Loss of Chd4 expression was observed in 56.4% of the GCs and 55.7% of the CRCs, and loss of Chd8 was observed in 35.7% of the GCs and 28.6% of the CRCs14. Finally, high levels of Snf2L expression in normal melanocytes contrast with undetectable expression in malignant melanoma. Depletion of Snf2L in HeLa cells led to enhanced proliferation and increased migration. These phenomena were explained by transcriptome profiling, which identified Snf2L as a modulator of the Wnt signaling network15. Thus, examples above show that the complete loss of particular chromatin remodeler expression is one of the way to support specific cancer cell.
Interaction with long non-coding RNA: There are mounting evidence, that interaction with RNA is another feature and possibility to control chromatin remodeling machines. Therefore it is not surprising that cancer cells use it to support uncotrolled cell division. Catalytic subunits of the SWI/SNF complex, Brg1 and Brm have been described to be regulated by lncRNA. For example, high expression of UCA1 (long non-coding RNA urothelial carcinoma associated 1) promotes bladder cancer cell proliferation by inhibiting chromatin remodeler Brg1. Depletion of UCA1 by RNAi resulted in upregulated p21 levels and inhibition of cell replication, while overexpressed UCA1 reduced p21 protein and promoted cell growth. UCA1 highly expressed tissue samples are often with Brg1 high expression and it was found that UCA1 impairs both binding of Brg1 to the p21 promoter and chromatin remodeling activity of Brg116. Another long noncoding RNA (lncRNA) called lncBRM is highly expressed in liver cancer stem cells (CSCs) and hepatocellular carcinoma (HCC) tumours. LncBRM is required for the self-renewal maintenance of liver CSCs and tumour initiation. In liver CSCs, lncBRM associates with Brm to initiate the Brg1/Brm switch and the Brg1-embedded BAF complex triggers activation of YAP1 (yes-associated protein 1) signalling. Moreover, expression levels of lncBRM together with YAP1 signalling targets are positively correlated with tumour severity of HCC patients17. It has been discussed in the literature, that the interaction between lncRNAs and the SWI/SNF complexes can be divided into two models in cancer: the binding model and the recruiting model. The binding model states that lncRNAs can directly bind to the subunit of the SWI/SNF complexes and serve as a guide to anchor the SWI/SNF complexes or function as a decoy to keep chromatin modifiers away from specific genomic sites. The recruiting model involves lncRNAs recruited SWI/SNF complexes or some core subunits to the target gene, thereby affecting the gene structure and expression18.
Overexpression: Overexpression without mutation of chromatin remodeler proteins is another way of how chromatin remodelers contribute to cancer. This group is of particular interest because of its promise for epigenetic therapy. For example, overexpression of Brg1 correlates with colon cancer progression through upregulating WNT3A expression. Knocking down Brg1 expression significantly suppressed the proliferation and invasion in colon cancer cells19. In breast cancer, Brg1 was overexpressed and upregulated de novo lipogenesis, which is crucial for cancer cell proliferation. Knockdown of Brg1 attenuated lipid synthesis by impairing the transcription of enzymes catalyzing fatty acid and lipid synthesis. Remarkably, exogenous addition of palmitate, the key intermediate in fatty acid synthesis, rescued the cancer cell proliferation defect caused by Brg1 knockdown20. Snf2H was overexpressed in human gliomas and contributed to glioma proliferation, invasion, and chemoresistance through regulation of cyclin E and NF-κB pathway, which is dependent on its interaction with Rsf-121. Snf2H was also overexpressed in breast cancers and contributed to cancer cell proliferation, invasion and correlated with poor prognosis. Snf2H overexpression correlated with Rsf-1 expression levels and downregulated cyclin A, MMP2 expression and upregulated p21 expression22. The expression of Ino80, the SWI/SNF ATPase of INO80 complex, was elevated in melanoma cells and patient melanomas. Ino80 silencing selectively inhibited melanoma cell proliferation, anchorage-independent growth, tumorigenesis, and tumor maintenance. Mechanistically, Ino80 occupies >90% of superenhancers, and its occupancy is dependent on transcription factors such as MITF and Sox9. Ino80 binding reduces nucleosome occupancy and facilitates Mediator recruitment, thereby promoting oncogenic transcription. Consistently, genes co-occupied by Ino80 and Med1 are selectively expressed in melanomas compared with melanocytes23. Ino80 was highly expressed in cervical cancer cell lines and tumor samples and promoted cell proliferation and tumorigenesis24. Ino80, is highly expressed in non-small-cell lung cancer (NSCLC) cells compared with normal lung epithelia cells. Its expression, as well as that of another subunit Ino80B, negatively correlates with disease prognosis in lung cancer patients. Functionally, Ino80 silencing inhibits NSCLC cell proliferation and anchorage-independent growth and tumor formation. It occupies enhancer regions near lung cancer-associated genes, and its occupancy correlates with increased genome accessibility and enhanced expression of downstream genes25. Baz2A, a factor previously implicated in epigenetic rRNA gene silencing, is overexpressed in prostate cancer and is involved in maintaining prostate cancer cell growth. Baz2A regulates numerous protein-coding genes and directly interacts with EZH2 to maintain epigenetic silencing at genes repressed in metastasis. Baz2A overexpression is tightly associated with a molecular subtype displaying a CpG island methylator phenotype26. Chormatin remodeler Lsh, furthered malignant progression of nasopharyngeal carcinoma (NPC) by modifying cancer cell metabolism to support epithelial-mesenchymal transition (EMT). Lsh expression was increased in NPC, where it was controlled by the Epstein-Barr virus-encoded protein LMP1. In NPC cells Lsh promoted cancer progression in part by regulating expression of fumarate hydratase (FH), a core component of the tricarboxylic acid cycle. Lsh bound to the FH promoter, recruiting the epigenetic silencer factor G9a to repress FH transcription. Clinically, it was found that the concentration of TCA (tricarboxylic acid) intermediates in NPC patient sera was deregulated in the presence of Lsh27. Thus, increased expression of chromatin remodelers in cancer is a wide and diverse group with its clinical potential as biomarker uses as well as therapeutics.
De Novo mutations in chromatin remodelers: These alterations contribute to cancer pathogenesis by deregulating chromatin remodelers. Whole-exome sequencing of Sézary syndrome tumor-normal sample pairs and mutation analysis identified somatic mutations in chromatin remodelers Brg1 and Chd328. Chromatin modifying genes including SETD2, Ino80 and ARID1B were commonly mutated in hepatosplenic T cell lymphoma, affecting 62% of cases29. Chd4 among other five proteins was idenified carrying the most relevant somatic oncogenic mutations in quadruple wild-type gastrointestinal stromal tumors is a genomic subgroup lacking KIT/PDGFRA/RAS pathways mutations, with an intact succinate dehydrogenase complex30. De novo mutations in chromatin remodeling complexes could possibly lead to structural or even functional changes, i.e. chromatin remodeling complexes are not properly formed and not targeted to the right loci (loss-of-function mutation) or targeted to another DNA loci (gain-of-function mutation).
Targeting chromatin remodelers to improve treatment efficiency in precision medicine
It is known that epigenetic regulation plays an essential role in tumor development and epigenetic modifiers are considered potential druggable candidates. For example, Chd4 was described as a potential pharmacological target to inhibit breast cancer growth. Chd4 silencing significantly reduced tumor growth and proliferation of MCF10DCIS.com cells (human breast cancer cell line, known for its capability of recapitulating the various stages of the malignancy when transplanted in an immune-compromised host). Chd4 depletion arrests proliferation by inducing a G0/G1 block of cell cycle associated with up-regulation of CDKN1A (p21)31. On the other hand, exsisting cancer therapies are not that efficient, some patients are developing chemoresistance, which is a major barrier to efficient chemotherapy. Recent reports show, that inhibiting specific chromatin remodelers actually improve treatment efficiencies. For example, gemcitabine is a standard chemotherapeutic agent for locally advanced and metastatic pancreatic cancer. However, some cells establish acquired chemoresistance and Brg1 knockdown can effectively reverse the chemoresistance to gemcitabine. Brg1 promotes both intrinsic and acquired chemoresistance of pancreatic cancer cells, and Brg1 crosstalks with Akt signalling to form a positive feedback loop to promote pancreatic cancer development32. Chd4 plays a crucial role in chemoresistance and the maintenance of stemness in liver cancer stem cells (CSCs) and its targeting suppresses the advanced hepatocellular carcinoma growth. Therefore, Chd4 was suggested as a novel therapeutic target in liver CSCs33. Moreover, inhibition of Chd4 has the potential to enhance the effectiveness of genotoxic agents used in acute meloid leukemia therapy34. However, loss of Chd4 confers cisplatin resistance and thereby modulates therapeutic response in BRCA2 mutant ovarian cancer cells. BRCA2 mutant ovarian cancers with reduced Chd4 expression significantly correlate with shorter progression-free survival and shorter overall survival35. Chemotherapies such as the topoisomerase II (TopoII) inhibitor etoposide effectively reduce disease in a minority of patients with non-small-cell lung cancer. A promising potential epigenetic target is the methyltransferase EZH2. EGFR and Brg1 mutations are genetic biomarkers that predict enhanced sensitivity to TopoII inhibitor in response to EZH2 inhibition. Brg1 loss-of-function mutant tumours respond to EZH2 inhibition with increased S phase, anaphase bridging, apoptosis and TopoII inhibitor sensitivity. Conversely, EGFR and Brg1 wild-type tumours upregulate Brg1 in response to EZH2 inhibition and ultimately become more resistant to TopoII inhibitor. EGFR gain-of-function mutant tumours are also sensitive to dual EZH2 inhibition and TopoII inhibitor, because of genetic antagonism between EGFR and Brg136. Moreover, ectopic expression of the Brg1-bromodomain (Brg1-BRD) construct inhibits γ-H2AX and DNA double-strand break repair after irradiation and increases the radiosensitivity in various human cancer cells, including HT29 colon cancer. Brg1-BRD radiosensitizes tumour cells by a dominant negative activity against Brg137. Thus, mountig evidence show the opportunity of targeting chromatin remodelers in precision medicine to treat the genetically complex diseases such as cancer.
Recently, epigenetic reader domains of chromatin remodelers arose as possible future targets in cancer therapy. Structures of bromodomains were solved38,39 and specific inhibitors Baz2-ICR, GSK2801 and Pfi-3 of bromodomains of Baz2A/Baz2B and Brg1 were created40–42. It was shown that exposure of embryonic stem cells to chemical probe Pfi-3 led to deprivation of stemness and deregulated lineage specification42. However, several studies reported that inhibition of bromodomain of Brg1 had no effect on cancer cell viability. Pfi-3 inhibitor was used in triple negative breast cancer cells and other cancer cells and did not influence the proliferation rate of the cancer cell lines. But then, specifically targeting the ATPase domain of Brg1 with ADAADiN (active DNA-dependent ATPase A domain inhibitor) molecule significantly decreased triple negative breast cancer cell proliferation43. GSK2801 seem to have excellent selectivity for Baz2A and Baz2B, with only low micromolar residual activity at BRD9 (bromodomain containing 9) and TAF1L (TATA-box binding protein associated protein like 1) and was shown to displace GFP-tagged Baz2A bromodomain from chromatin41. However, the effect of GSK2801 on various cancer cell proliferation remain unknown.
Conclusion
Deregulated chromatin remodelers arose as additional players in the cellular malignancy. There are mounting evidence that variety of cancers employ chromatin remodelers: they can be downregulated/lacking functional protein, increased expression, carry somatic mutations or support cancer through the interaction with long noncoding RNA. In parallel, the promise of epigenetic therapies is growing. It seems that targeting deregulated chromatin remodeling machines is additional level of personalizied medicine and has a promise to increase the efficiency of cancer treatment. Thus, further understanding the mechanisms how chromatin remodelers act in cancer and exploring their pharmaceutical potential might lead to more efficient therapies.
Acknowledgement
Apologies to all colleagues whose work could not be cited due to space limitations. Work of Laura Manelyte is funded by the DFG. Funding for open access charge: Regensburg University Library.
References
- Längst G, Manelyte L. Chromatin Remodelers From Function to Dysfunction. Genes Basel. 2015; 6: 299–324.
- Clapier CR, Cairns BR. The Biology of Chromatin Remodeling Complexes. Annu Rev Biochem. 2009; 78: 273–304.
- Flaus A, Martin DMA, Barton GJ, et al. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 2006; 34: 2887–905.
- Versteege I, Sévenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998; 394: 203–206.
- Witkowski L, Carrot Zhang J, Albrecht S, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary hypercalcemic type. Nat Genet. 2014; 46: 438–43.
- Conlon N, Silva A, Guerra E, et al. Loss of SMARCA4 Expression Is Both Sensitive and Specific for the Diagnosis of Small Cell Carcinoma of Ovary Hypercalcemic Type. Am J Surg Pathol. 2016; 40: 395–403.
- Clarke BA, Witkowski L, Ton Nu TN, et al. Loss of SMARCA4 (BRG1) protein expression as determined by immunohistochemistry in small-cell carcinoma of the ovary, hypercalcaemic type distinguishes these tumours from their mimics. Histopathology. 2016; 69: 727–738.
- Ramos P, Karnezis AN, Craig DW, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet. 2014; 46: 427–9.
- Karnezis AN, Wang Y, Ramos P, et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. J Pathol. 2016; 238: 389–400.
- Jelinic P, Schlappe BA, Conlon N, et al. Concomitant loss of SMARCA2 and SMARCA4 expression in small cell carcinoma of the ovary hypercalcemic type. Mod Pathol. 2016; 29: 60–66.
- Medina PP, Romero OA, Kohno T, et al. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat. 2008; 29: 617–622.
- Song S, Walter V, Karaca M, et al. Gene Silencing Associated with SWI/SNF Complex Loss during NSCLC Development. Mol Cancer Res. 2014; 12.
- Ramalingam P, Croce S, McCluggage WG. Loss of expression of SMARCA4 (BRG1), SMARCA2 (BRM) and SMARCB1 (INI1) in undifferentiated carcinoma of the endometrium is not uncommon and is not always associated with rhabdoid morphology. Histopathology. 2017; 70: 359–366.
- Kim MS, Chung NG, Kang MR, et al. Genetic and expressional alterations of CHD genes in gastric and colorectal cancers. Histopathology. 2011; 58: 660–668.
- Eckey M, Kuphal S, Straub T, et al. Nucleosome remodeler SNF2L suppresses cell proliferation and migration and attenuates Wnt signaling. Mol Cell Biol. 2012; 32: 2359–71.
- Wang X, Gong Y, Jin B, et al. Long non-coding RNA urothelial carcinoma associated 1 induces cell replication by inhibiting BRG1 in 5637 cells. Oncol Rep. 2014; 32: 1281–90.
- Zhu P, Wang Y, Wu J, et al. LncBRM initiates YAP1 signalling activation to drive self-renewal of liver cancer stem cells. Nat Commun. 2016; 7: 13608.
- Tang Y, Wang J, Lian Y, et al. Linking long non-coding RNAs and SWI/SNF complexes to chromatin remodeling in cancer. Mol Cancer. 2017; 16: 42.
- Lin S, Jiang T, Ye L, et al. The chromatin-remodeling enzyme BRG1 promotes colon cancer progression via positive regulation of WNT3A. Oncotarget. 2016; 7: 86051–86063.
- Wu Q, Sharma S, Cui H, et al. Targeting the chromatin remodeling enzyme BRG1 increases the efficacy of chemotherapy drugs in breast cancer cells. Oncotarget. 2016; 7: 27158–75.
- Zhao XC, An P, Wu XY, et al. Overexpression of hSNF2H in glioma promotes cell proliferation, invasion, and chemoresistance through its interaction with Rsf-1. Tumor Biol. 2016; 37: 7203–7212.
- Jin Q, Mao X, Li B, et al. Overexpression of SMARCA5 correlates with cell proliferation and migration in breast cancer. Tumor Biol. 2015; 36: 1895–1902.
- Zhou B, Wang L, Zhang S, et al. INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev. 2016; 30: 1440–53.
- Hu J, Liu J, Chen A, et al. Ino80 promotes cervical cancer tumorigenesis by activating Nanog expression. Oncotarget. 2016; 7: 72250–72262.
- Zhang S, Zhou B, Wang L, et al. INO80 is required for oncogenic transcription and tumor growth in non-small cell lung cancer. Oncogene. 2016. doi:10.1038/onc.2016.311
- Gu L, Frommel SC, Oakes CC, et al. BAZ2A (TIP5) is involved in epigenetic alterations in prostate cancer and its overexpression predicts disease recurrence. 2014; Nat Genet. 47: 22–30.
- He X, et al. Molecular and Cellular Pathobiology Chromatin Remodeling Factor LSH Drives Cancer Progression by Suppressing the Activity of Fumarate Hydratase. Cancer Res. 76: 5743–55.
- Carolina Da Silva Almeida A, Abate F, Khiabanian H, et al. The mutational landscape of cutaneous T-cell lymphoma and Sézary syndrome. doi:10.1038/ng.3442
- McKinney M, Moffitt AB, Gaulard P, et al. The Genetic Basis of Hepatosplenic T Cell Lymphoma. Cancer Discov. 2017.
- Pantaleo MA, Urbini M, Indio V, et al. Genome-wide Analyses Identifies MEN1 and MAX Mutations and a Neuroendocrine-like Molecular Heterogeneity in Quadruple WT GIST. Mol Cancer Res. 2017.
- D’Alesio C, Punzi S, Cicalese A, et al. RNAi screens identify CHD4 as an essential gene in breast cancer growth. Oncotarget. 2016; 7: 80901–80915.
- Liu X, Tian X, Wang F, et al. BRG1 promotes chemoresistance of pancreatic cancer cells through crosstalking with Akt signalling. Eur J Cancer. 2014; 50: 2251–2262.
- Nio K, Yamashita T, Kaneko S. Chromodomain-helicase-DNA-binding protein 4: a novel therapeutic target in liver cancer stem cells. Chinese Clin Oncol. 2016; 0.
- Sperlazza J, Rahmani M, Beckta J, et al. Depletion of the chromatin remodeler CHD4 sensitizes AML blasts to genotoxic agents and reduces tumor formation. doi:10.1182/blood-2015-03
- Guillemette S, Serra RW, Peng M, et al. Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 2015; 29: 489–94.
- Fillmore CM, Xu C, Desai PT, et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature. 2015; 520: 239–42.
- Kwon SJ, Lee SK, Na J, et al. Targeting BRG1 Chromatin Remodeler via Its Bromodomain for Enhanced Tumor Cell Radiosensitivity In Vitro and In Vivo. Mol. Cancer Ther. 2015; 14.
- Filippakopoulos Pk, Picaud S, Mangos M, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012; 149: 214–31.
- Tallant C, Valentini E, Fedorov O, et al. Molecular basis of histone tail recognition by human TIP5 PHD finger and bromodomain of the chromatin remodeling complex NoRC. Structure. 2015; 23: 80–92.
- Drouin L, McGrath S, Vidler LR et al. Structure enabled design of BAZ2-ICR, a chemical probe targeting the bromodomains of BAZ2A and BAZ2B. J Med Chem. 2015; 58: 2553–9.
- Chen P, Chaikuad A, Bamborough P, et al. Discovery and Characterization of GSK2801, a Selective Chemical Probe for the Bromodomains BAZ2A and BAZ2B. doi:10.1021/acs.jmedchem.5b00209
- Fedorov O, Castex J, Tallant C, et al. Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance. Sci Adv. 2015; 1: e1500723.
- Wu Q, Madany P, Dobson JR, et al. The BRG1 chromatin remodeling enzyme links cancer cell metabolism and proliferation. Oncotarget. 2016; 7: 38270–38281.