
Commentary: p62 plays a protective role in the autophagic degradation of polyglutamine protein oligomers in polyglutamine disease model flies
Yuji Saitoh1,2 and Yoshitaka Nagai1,3*
1Department of Degenerative Neurological Diseases, National Institute of Neuroscience, National Center of Neurology and Psychiatry, 4-1-1 Ogawa-Higashi, Kodaira, Tokyo 187-8502, Japan
2Department of Neurology, National Center Hospital, National Center of Neurology and Psychiatry, 4-1-1 Ogawa-higashi, Kodaira, Tokyo 187-8551, Japan
3Department of Neurotherapeutics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan
In most neurodegenerative diseases, such as Alzheimer disease (AD), Parkinson disease, polyglutamine (polyQ) diseases, and amyotrophic lateral sclerosis (ALS), deposition of various proteins in the brain are common pathological features. These pathogenic proteins are prone to be misfolded, form oligomers and aggregates, and eventually accumulate inside the affected neurons as inclusion bodies, this tendency is accelerated by pathogenic mutations linked to familial forms of these diseases. In the pathomechanisms of these neurodegenerative diseases, recent studies suggested that intermediate structures, including monomers or oligomers among the aggregation cascade of these pathogenic proteins could be more toxic for neurons1. Therefore, clearance of these proteins, especially more toxic oligomeric species, should be one of the promising therapeutic strategies against these intractable neurodegenerative diseases. The two major cellular degradation systems, the ubiquitin-proteasome system (UPS) and the autophagy-lysosome system (hereafter called autophagy), are thought to be involved in the degradation of these pathogenic proteins, although the precise mechanisms have not been fully understood2. The UPS could degrade the precursor monomeric proteins but may be inadequate for degrading aggregates of these pathogenic proteins efficiently because aggregated proteins are difficult to be unfolded and to be delivered into the proteasome. On the other hand, autophagy can degrade even large protein oligomers by sequestering them into the autophagosome. Because these processes accompany the non-specific degradation of other cellular molecules in the cytosol as well, autophagy is considered as a non-selective, bulk degradation system. Recently, mounting evidence indicates the existence of selective autophagy, which is mediated via the adaptor molecules recognizing both target proteins and autophagosome constituent proteins3. The p62/sequestosome 1 protein (hereafter called p62) is considered as an adaptor molecule for selective autophagy that degrades the ubiquitinated proteins, because p62 has binding domains for both ubiquitinated proteins and autophagosomes4,5. In the brain of neurodegenerative diseases, p62 is found to be co-localized with ubiquitinated intraneuronal inclusions consisting of the pathogenic proteins, indicating some sort of association between p62 and these toxic proteins. However, whether p62 recognizes these pathogenic proteins as substrates for p62-mediated selective autophagy and plays a role in the pathogenesis of neurodegenerative diseases in vivo has remained unclear.
In our recent article6, we explored the role of p62 in the pathologic conditions and demonstrated that p62 plays an important role in the autophagic degradation of polyQ protein, especially its oligomers (Figure 1). We also showed a protective role of p62 in various neurodegenerative disease models as well. We used Drosophila models as neurodegenerative disease in vivo models, taking advantage of their suitability for genetic analysis. In the polyQ model flies expressing a truncated form of the mutant MJD protein with an expanded Q78 repeat in the eye, which exhibit polyQ protein aggregates and eye degeneration, p62 was found to be co-localized with cytoplasmic polyQ protein aggregates. The genetic ablation of p62 causes exacerbation of eye degeneration accompanied by the enhanced accumulation of cytoplasmic polyQ protein aggregates, indicating that p62 has the protective role against polyQ-induced degeneration. For the mechanistic insight for these results, we obtained two important results. First, genetic ablation of autophagy-associated genes showed the exacerbation of polyQ-induced degeneration accompanied by the enhancement of cytoplasmic polyQ protein aggregates, similar to genetic ablation of p62. Second, knockdown of autophagy-associated gene together with p62 knockdown did not show any additive effects on polyQ-induced degeneration, suggesting that p62 plays a protective role against polyQ protein toxicity via autophagy. On the contrary, genetic ablation of UPS-associated gene concomitantly with p62 knockdown further exacerbated polyQ-induced degeneration and polyQ protein aggregates, suggesting that the protective role of p62 against polyQ protein is different from that of UPS. Furthermore, by the biochemical chase analyses using the inducible polyQ protein expression models, we clarified the protective effect of p62 against the polyQ protein is dependent on the autophagic degradation of the polyQ proteins, especially its oligomeric species. Interestingly, we found that p62 exerts its protective effects not only against the polyQ protein but also against another pathogenic protein associated with neurodegenerative disease, such as the TDP-43 protein related to ALS, although no protective effect was observed against the tau protein and the amyloid-β (Aβ) peptides. Our research reveals the important aspects that p62 plays a protective role in neurodegenerative diseases and its effect exerts via autophagic degradation of the pathogenic protein, indicating p62 is expected to be targeted as a general therapeutic strategy against protein-misfolding neurodegenerative diseases in the future.
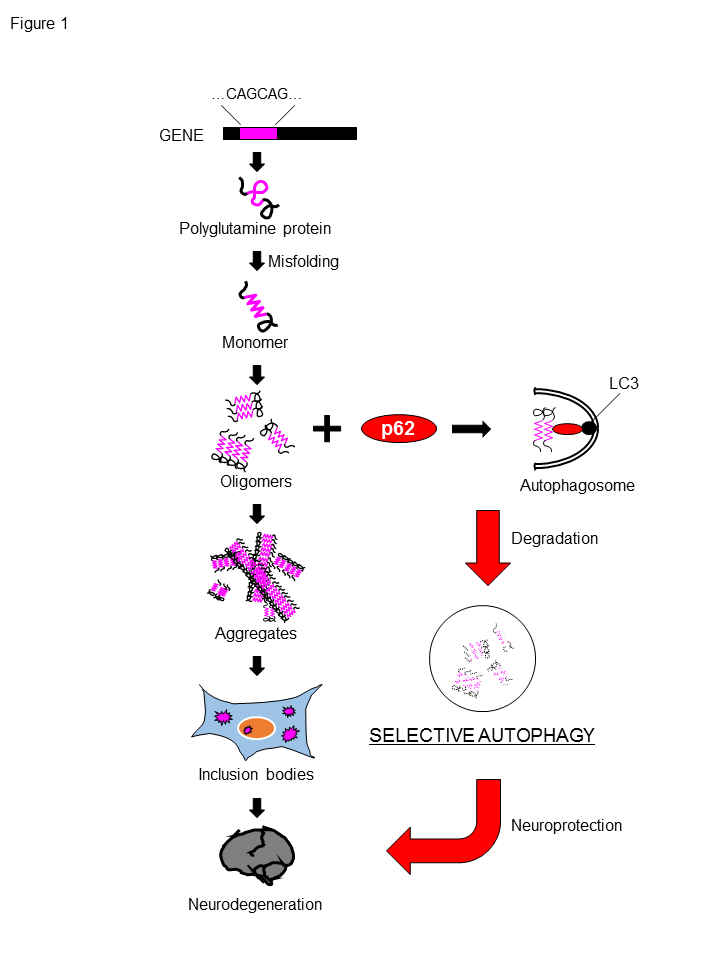
Figure 1. p62 degrades polyglutamine protein oligomers via autophagy.
The expansion of a CAG repeat encoding for polyQ stretch leads to synthesis of polyQ proteins, which are prone to be misfolded, form oligomers and aggregates. and eventually accumulate as inclusion bodies in neuron. p62-mediated selective autophagy can degrade polyQ proteins, especially its oligomers, which are considered to be more toxic to neurons, resulting in the neuroprotection against toxicity of polyQ protein.
However, our research presents several limitations. In our paper, we could not clearly demonstrate why p62 plays a protective role against the polyQ protein and the TDP-43 protein, but not against the tau protein and the Aβ peptide. We speculated that since the tau protein is degraded by the UPS rather than autophagy, and the Aβ peptides are known to be degraded by endopeptidase, p62 knockdown could not show any effects on these pathogenic agents7-11. Another limitation is that we could not reveal how p62 recognizes the pathogenic proteins such as the polyQ proteins. The recognition of these pathogenic proteins could be mediated via the ubiquitination of these proteins and their affinities for p62. In this regards, Matsumoto et al. revealed that phosphorylation of p62 at S405 by casein kinase 2 may regulate the clearance of polyQ proteins12. Lim et al. also showed that autophagy-related kinase ULK1 phosphorylates p62 at S409, resulting in the enhancement the autophagic degradation of the polyQ proteins13. These reports indicate that the post-translational modifications of p62 could also be one of the targets applying p62 as therapeutic options. On the other hand, p62 plays a role in the several signaling pathways, such as the Keap1-Nrf2 pathway or the NK-kB pathway. Because several reports revealed that these pathways are associated with polyQ protein toxicity14,15, the regulation of p62 could also contribute to ameliorate polyQ protein toxicity via these pathways, in addition to p62-mediated selective autophagy. Further studies are required to develop the efficient therapeutics for these intractable neurodegenerative diseases.
Fundings
This work was supported in part by Grants-in-Aid for Scientific Research (B) to Y.N. from the Japan Society for the Promotion of Science (JSPS), Japan; by Grants-in-Aid for Scientific Research on Priority Areas (Proteolysis to Y.N.) and on Innovative Areas (Synapse and Neurocircuit Pathology to Y.N.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; by Health Labour Sciences Research Grants for Research on Development of New Drugs and the Research Committee for Ataxic Diseases to Y.N. from the Ministry of Health, Labour and Welfare, Japan; by a grant from Core Research for Evolutional Science and Technology (CREST) of the Japan Science and Technology Agency to Y.N.
References
- Nagai Y, Inui T, Popiel HA, et al. A toxic monomeric conformer of the polyglutamine protein. Na Struct Mol Biol. 2007; 14: 332-40.
- Rubinsztein, DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006; 443: 780-86.
- Johansen T, and Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7: 279-96.
- Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007; 282: 24131-45.
- Ichimura Y, Kumanomidou T, Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008; 283: 22847-57.
- Saitoh Y, Fujikake N, Okamoto Y, et al. p62 plays a protective role in the autophagic degradation of polyglutamine protein oligomers in polyglutamine disease model flies. J Biol Chem. 2015; 290: 1442-53.
- Petrucelli L, Dickson D, Kehoe K, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004; 13: 703-14.
- Shimura H, Schwartz D, Gygi SP, et al. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem. 2004; 279: 4869-76.
- Lee MJ, Lee JH, and Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol. 2013; 105: 49-59.
- Iwata N, Tsubuki S, Takaki Y, et al. Identification of the major Aβ1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000; 6: 143-50.
- Qiu WQ, Walsh DM, Ye Z, et al. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J Biol Chem. 1998; 273: 32730-8.
- Matsumoto G, Wada K, Okuno M, et al. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 2011; 44: 279-89.
- Lim J, Lachenmayer ML, Wu S, et al. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015; 11: e1004987.
- Bott LC, Badders NM, Chen KL, et al. A small-molecule Nrf1 and Nrf2 activator mitigates polyglutamine toxicity in spinal and bulbar muscular atrophy. Hum Mol Genet. 2016; 25: 1979-89.
- Marcora E, Kennedy MB. The Huntington's disease mutation impairs Huntingtin's role in the transport of NF-κB from the synapse to the nucleus. Hum Mol Genet. 2010; 19: 4373-84.