
Identification of a novel MTTP splice variant c.394-2A>C in an infant with abetalipoproteinemia
Dinesha M. Vidanapathirana1,2*, Eresha Jasinge1, Samantha Waidyanatha3, Amanda J. Hooper4,5, John R. Burnett4,5
1Department of Chemical Pathology, Lady Ridgeway Hospital for Children, Sri Lanka
2Department of Pathology, Faculty of Medical Sciences, University of Sri Jayewardenepura, Sri Lanka
3Department of Paediatrics, Lady Ridgeway Hospital, Sri Lanka
4Department of Clinical Biochemistry, PathWest Laboratory Medicine, Royal Perth Hospital and Fiona Stanley Hospital Network, Perth, Australia
5School of Medicine, University of Western Australia, Australia
Abstract
Abetalipoproteinemia (ABL) is a rare autosomal recessive disorder of lipoprotein metabolism caused by mutations in the microsomal triglyceride transfer protein (MTTP) gene. To date, less than 100 cases of ABL have been reported worldwide. It is characterized biochemically by the absence or extremely low levels of low-density lipoproteins in the blood. We report a four-month-old girl, born to consanguineous parents, who presented with steatorrhea, failure to thrive, marked hypolipidemia and acanthocytosis, with a similar history having been noted in her older sibling. DNA sequencing revealed the infant to be homozygous for a novel pathogenic MTTP splice variant c.394-2A?C. Family screening revealed her sister to be homozygous for the same MTTP variant while her parents were heterozygotes. Early diagnosis and treatment of ABL in the form of a low-fat diet and fat-soluble vitamin supplementation can mitigate neuropathy and retinopathy. We believe that this is the first identification of an infant with a novel mutation for abetalipoproteinemia in Sri Lanka.
Introduction
Abetalipoproteinemia (ABL; OMIM 200100) is a very rare autosomal recessive disorder of lipoprotein metabolism.1,2 It is caused by mutations of the microsomal triglyceride transfer protein (MTTP) gene,3 the protein product of which is responsible for the transfer of cholesteryl esters, triglycerides and phospholipids onto the nascent apolipoprotein B (apoB), the structural component of intestinally-derived chylomicrons and hepatically-derived very low density lipoproteins (VLDL).4,5 Deficiency of MTTP results in absence of these apoB-containing lipoproteins and their remnants.4,5
ABL usually presents in infancy with failure to thrive, severe diarrhea and fat malabsorption.1,2 Patients also exhibit extremely low plasma lipid levels, acanthocytosis on peripheral blood film and, if untreated, may develop ataxic neuropathy and retinopathy later in childhood due to fat-soluble vitamin deficiency.3 We report an infant with ABL due to homozygosity for a novel pathogenic MTTP variant, c.394-2A?C, who presented with failure to thrive due to steatorrhea.
Case Report
A four-month-old Sri Lankan girl presented to our pediatric service for evaluation of failure to thrive, having been exclusively breastfed. She was the third child born by vaginal delivery to consanguineous parents after an uneventful pregnancy with a birth weight of 2.6 kg. There was no history of vomiting, pulmonary complaints, feeding difficulties, or recurrent infections. Her stools were described as being “greasy” from age six weeks. On clinical examination, the infant, although small and thin, appeared alert and in no distress. There were no dysmorphic features or cutaneous lesions. A cardiorespiratory examination was normal. She had no abdominal distension, tenderness or organomegaly. Fundoscopy showed no cataracts or retinal pigmentary changes. A neuromuscular examination was normal.
Laboratory examination revealed normal plasma electrolytes, glucose, urea and creatinine, with an isolated increase in AST at 82 U/L (<40) noted on liver function tests. A full blood count showed a hemoglobin of 9.1 g/dL (9.5–14.5), with normal white blood cell and platelet counts. A peripheral blood film revealed marked acanthocytosis. Stool analysis showed the presence of fat globules consistent with steatorrhea. A lipid profile revealed marked hypocholesterolemia (Table 1). An abdominal ultrasound scan showed an incidental 6 mm simple cyst in left adenexal region.
The patient’s parents, who were first cousins, were asymptomatic with essentially normal lipid levels (Table 1, Figure 1). Her 10-year-old sister, with mild learning difficulties and urolithiasis, was diagnosed biochemically with ABL at 2.5 years of age and was on a low-fat diet with fat-soluble vitamin supplements. A brother died at 1 year of age having been diagnosed biochemically with ABL at 6 months of age. DNA was isolated from peripheral blood and the sample was analyzed using massive parallel sequencing of lipid genes with targeted analysis of monogenic hypocholesterolemia genes (APOB, MTTP, PCSK9, SAR1B, ANGPTL3). The patient was found to be homozygous for a novel MTTP variant, NM_000253.3:c.394-2A?C, predicted to be pathogenic as classified by the 2015 American College of Medical Genetics and Genomics guidelines. No potentially pathogenic variants were identified in the other hypocholesterolemia genes. Family screening showed that her sister is also homozygous for the same MTTP variant, while the mother and father are heterozygotes.
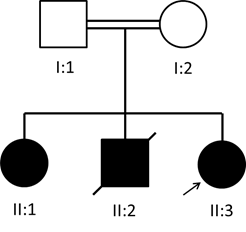
Fig. 1: Pedigree of the abetalipoproteinemia family
Table 1. Lipid profiles of the proband and her nuclear family.
| Analyte | Proband (II:3) 4 months |
Sister (II:1) 10 years |
Mother (I:2) 34 years |
Father (I:1) 43 years |
|---|---|---|---|---|
| Total Cholesterol (mg/dL) | 48 (112 - 200) |
41 (125 - 205) |
229 (133 - 227) |
245 (142 - 258) |
| Triglycerides (mg/dL) | 19 (34 - 112) |
12 (39 - 120) |
48 (40 - 163) |
118 (46 - 253) |
| LDL-cholesterol (mg/dL) | 13 | 11 (68 - 136) |
167 (70 - 156) |
167 (87 - 186) |
| HDL-cholesterol (mg/dL) | 32 | 31 (37 - 70) |
53 (36 - 77) |
55 (27 - 67) |
Reference intervals shown in parentheses.
The infant received dietary and nutritional advice and was commenced on fat-soluble vitamin (A, D, E, and K), as well as iron and folic acid supplementation.
Discussion
ABL is a rare inborn error of lipoprotein metabolism that was first described by Bassen and Kornzweig in 1950.6 Salt and colleagues subsequently named the disorder “a-beta-lipoproteinemia”.7 However, it was not until 1993 that mutations in MTTP were identified as the genetic basis of ABL.3 Since then, about 40 mutations in the MTTP gene have been identified.
Children with ABL are normal at birth, but present early in life with malabsorption of fat and fat-soluble vitamins leading to steatorrhea and failure to thrive and, if untreated, may progress to multisystem involvement.8 Hematological manifestations, as observed in our patient, include acanthocytosis and anemia.2
A similar clinical picture to ABL can be seen in homozygous familial hypobetalipoproteinema (FHBL), an autosomal codominant disorder due to mutations in APOB.2 In both conditions, LDL-cholesterol levels and apoB are absent or extremely low. However, obligate heterozygous parents of homozygous FHBL patients usually have approximately one third normal plasma levels of apoB and LDL-cholesterol, while obligate heterozygote parents of ABL patients have normal plasma lipoprotein profiles.9 Our proband’s parents were normolipidemic, consistent with a diagnosis of ABL (Table 1).
Genetic studies revealed that the proband was homozygous for pathogenic MTTP variant c.394-2A?C. This mutation has not been previously described and is absent from population databases.10 It is predicted to abolish the intron 4 splice acceptor site of the MTTP gene, likely resulting in a null variant. Other MTTP splicing mutations have previously been described in abetalipoproteinemia.11,12 The sister of our proband had a lipid profile similar to the proband and she was also found to be homozygous for MTTP c.394-2A>C. As expected, the parents were heterozygous carriers of the mutation.
The cornerstone of treatment is life-long dietary modification and the replacement of fat-soluble vitamins.13,14 A diet and minimal consumption of long chain fatty acids eliminates steatorrhea and allows for absorption of other nutrients essential for growth and development. Early diagnosis and treatment in the form of a low-fat diet and replacement of fat-soluble vitamins can mitigate neuropathy and retinopathy.13,14
In summary, we report an infant with ABL due to homozygosity for a novel pathogenic MTTP variant, c.394-2A?C who presented with failure to thrive due to steatorrhea.
Acknowledgement
We thank Ms Lan Nguyen for assistance with DNA sequencing.
References
-
- Burnett JR, Hooper AJ, Hegele RA. Abetalipoproteinemia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019.
- Zamel R, Khan R, Pollex RL, et al. Abetalipoproteinemia: two case reports and literature review. Orphanet J Rare Dis. 2008; 3(1): 19.
- Sharp D, Blinderman L, Combs KA, et al. Cloning and gene defects in microsomal triglyceride transfer protein associated with abetalipoproteinaemia. Nature. 1993; 365: 65-9.
- Berriot-Varoqueanux N, Aggerbeck LP, Samson-Bouma ME, et al. The role of the triglyceride transfer protein in abetalipoproteinemia. Annu Rev Nutr. 2000; 20: 663-97.
- Hooper AJ, Burnett JR, Watts GF. Contemporary aspects of the biology and therapeutic regulation of microsomal triglyceride transfer protein. Circ Res. 2015; 116(1): 193-205.
- Bassen FA, Kornzweig AL. Malformation of the erythrocytes in a case of atypical retinitis pigmentosa. Blood. 1950; 5: 381-7.
- Salt HB, Wolff OH, Lloyd JK, et al. On having no beta-lipoprotein: A syndrome comprising a-beta-lipoproteinaemia, acanthocytosis, and steatorrhoea. Lancet. 1960; 2: 325-9.
- Rahalkar AR, Hegele RA. Monogenic pediatric dyslipidemias: Classification genetics and clinical spectrum. Mol Genet and Metab. 2008; 93: 282-94.
- Burnett JR, Hooper AJ, Bell DA, et al. Clinical utility gene card for: abetalipoproteinaemia – Update 2014. Eur J Hum Genet. 2015; 23(6). doi: 10.1038/ejhg.2014.224.
- ExAC (Exome Aggregation Consortium). http://exac.broadinstitute.org/
- Hammer MB, El Euch-Fayache G, Nehdi H, et al. Clinical features and molecular genetics of two Tunisian families with abetalipoproteinemia. J Clin Neurosci. 2014; 21: 311-5.
- Ohashi K, Ishibashi S, Osuga J, et al. Novel mutations in the microsomal triglyceride transfer protein gene causing abetalipoproteinemia. J Lipid Res. 2000; 41: 1199-204.
- Hooper AJ, Burnett JR. Chapter 37. Abetalipoproteinemia and hypobetalipoproteinemia. In, Hollak CEM, Lachman R, Sedel F, editors. Inherited Metabolic Diseases in Adults: A Clinical Guide, Oxford Press, 2016; 225-8.
- Ng DM, Burnett JR, Bell DA, et al. Update on the diagnosis, treatment and management of rare genetic lipid disorders. Pathology. 2019; 51: 193-201.