
Pulmonary Alveolar Proteinosis: Own Experience of Diagnosis and Treatment
Boris M. Ariel1,2*, Ivetta V. Dvorakovskaya1,3, Ludmila N. Novikova3, Mikhail M. Ilkovich3
1St. Petersburg Research Institute of Phthisiopulmonology, Health Ministry of Russia, St. Petersburg, Russia
2St. Petersburg City Mortem Bureau, St. Petersburg, Russia
3Pulmonology Clinic of Pavlov State Medical University, St. Petersburg, Russia
Abstract
Pulmonary alveolar proteinosis (PAP) is a rare interstitial lung disease with severe impairment of respiratory function caused by some genetic abnormalities of surfactant production and utilization. One of the clue moments in the pathogenesis of this disease which can lead to respiratory failure and death is the pulmonary fibrosis development occurring occasionally in patients with moderate and severe PAP. According to our own experience in 1977-2018 whole and segmental lung lavage remains the first-line treatment of PAP; 70 patients (82%) had shown an improvement after this treatment. A noticeable improvement was achieved in 67 patients (79%), and no serious complications were observed. The 5-year survival rate reached 100%. Nevertheless, delayed diagnosis and incorrect administration of antibiotics and tuberculostatics reduce the probability of a long symptom-free period after lung lavage and resolution of the disease.
Pulmonary alveolar proteinosis, or alveolar lipoproteinosis (PAP) is a diffuse lung disease that results from the accumulation of lipoproteinaceous material in the alveoli and alveolar macrophages due to abnormal surfactant homeostasis. Identification of the granulocyte-macrophage colony-stimulating factor (GM-CSF) as an indispensable mediator of macrophage maturation and surfactant catabolism was the key discovery leading to the current understanding of the pathogenesis of most forms of PAP. Impaired GM-CSF bioavailability due to anti-GM-CSF autoimmunity is the cause of approximately 90% of adult PAP cases. Abnormal macrophage function due to endogenous or exogenous triggers, GM-CSF receptor defects, and other genetic abnormalities of surfactant production account for the remainder of causes. It would be impossible not to agree with ?.Kumar et al.1 that this is the case.
According to the recent years' literature, difficulties in the diagnosis of pulmonary alveolar proteinosis remain, despite of the fact that a diagnosis involved the latest research methods2-4. This is an opportunity to discuss a number of key questions of diagnosis and treatment of this lung pathology again, using our new observations, in comparison with the results that were published earlier.
First described by S. H. Rosen et al.5, PAP is a rare pathology of the lungs and occurs with a frequency of 3.7 per 1000000 adults6. In most cases, patients are the younger people mainly of 20-50 years, and the men are affected 2-3 times more often than the women. The predominance of smokers (about 70%) is noted7. In 90% of the cases PAP is considered as idiopathic and in 5-10% it develops in individuals with immunosuppression therapy in cancer, including blood diseases8-12, after the lung transplantation, in infectious diseases13, 14, after inhalation of silicon, aluminum or titanium dust that is proved in animal experiments15-17. In rare cases PAP was observed in children with absence of the thymus, with reduced production of immunoglobulin A, or in the presence of any other immune deficiency18.
In recent years, an idea was developed that PAP is an autoimmune pathology1, 19-21. Thus, in serum and in broncho-alveolar lavage fluid (BAL) of patients with PAP there were identified autoantibodies against granulocyte-macrophage colony-stimulating factor (GM-CSF). Consequently, it disrupts the process of reutilization of surfactant with ? type alveolocytes and alveolar macrophages, and it is accumulated in the alveoli in excess, reducing the surface of the gas exchange and alveolar clearance.
Since clinical and radiological features of PAP are nonspecific, the diagnosis verification is provided by the histological examination of the lungs. A distinguishing characteristic is considered to be positive PAS-reaction, that is purple red protein and lipid staining of alveolar content under the influence of Schiff reagent22, 23 in association with negative alcian blue and mucicarmine staining. PAS-positive substance in the alveoli is stained with oil red O and sudan black for fat in frozen and paraffin sections, and immunohistochemically - with antibodies against surfactant apoprotein. Then, in presence of characteristic microscopic picture, the diagnosis of PAP is confirmed in ex juvantibus treatment with GM-CSF24 and other methods including therapeutic broncho-alveolar lavage25-27. In addition, there are some other alternative or complementary treatments, namely plasmapheresis, recombinant GM-CSF, rituximab (a drug containing a synthetic monoclonal antibody to CD20 antigen of B-lymphocytes), or stem cell transplants28. M.Luisetti et al.29 showed that after plasmapheresis the condition of patients improves, the titer of autoantibodies against GM-CSF reduces and the time interval till the repeated therapeutic broncho-alveolar lavage increases.
Being in effect benign, the lung damage in PAP progresses very slowly, and the therapeutic prognosis is generally favourable. 5-year survival rate is around 75%21, 30. In the case of progressing PAP as in many other chronic lung diseases a severe respiratory distress, a secondary pulmonary hypertension, a "pulmonary heart," deformed fingers ("fingers of Hippocrates") develop. In addition, there are known other complications such as bacterial or fungal infections23.
Given the recent advances in the study of the PAP pathogenesis, many scientists came to the conclusion that PAS-positive, diastase-resistant staining of the smear of BAL is enough to confirm the diagnosis of PAP. Detection of antibodies to GM-CSF in serum and in BAL is of the equal importance1.
Results
In 1977-2018 we observed 85 cases of pulmonary alveolar proteinosis. 59 patients (69%) were male. Their mean age was 38± 9.8 years. 60 patients (71%) were smokers. In the anamnesis of 47 patients (55%) there were indications of the long-term work with acids, alkalis, gasoline.
Most patients complained of breath shortness and cough (respective 59 (69%) and 54 (64%). Sometimes they have weakness (18, - 21%), weight loss (10, - 12%) and low-grade fever (10, - 12%). In 2 patients (2%) hemoptysis was periodically observed (table 1).
Table 1. Symptoms and signs in patients with PAP before broncho-alveolar lavage (N=85)
| Symptoms | Number of patients |
|---|---|
| Dyspnea | 59 |
| Cough | 54 |
| Fatigue | 18 |
| Low grade fever | 10 |
| Weight loss | 10 |
| Hemoptysis | 2 |
| Asymptomatic | 21 |
Functional impairment of lungs was mild in most of the patients. There was a trend towards the formation of a restrictive syndrome with the progression of the disease. Increase in systolic blood pressure to an average of 30.8±14,8 mm Hg in the pulmonary artery was noted in 23 patients (27%).
Lung changes detected in X-ray and HRCT studies allowed to suspect the PAP with high probability. At the same time, the correlation between radiological and clinical data were missing, and pronounced radiographic changes were accompanied in some cases by scarce clinical symptoms. In advanced stages of the lung disease widespread fibrosis was marked.
In 21 patients (25%) the disease was asymptomatic, and changes in the lungs were detected at prophylactic fluorographic studies.
The period of time from the first symptoms to biopsy taking was 4-92 (average 34) months, which is much greater than that of European authors, according to which this period is 1 to 18 (average 7) months21.
Before the correct diagnosis of PAP every fourth patient was diagnosed with either pneumonia, tuberculosis, or sarcoidosis, in several rare cases - Langerhans' cells histiocytosis, idiopathic fibrosing alveolitis, and amyloidosis.
The diagnosis of "double pneumonia" caused the administration of the massive antibacterial therapy for the duration of 1 month (on average), and sometimes much longer (up to 10 months).
As a rule, after a long period of unsuccessful pneumonia treatment, the diagnosis was revised in favor of tuberculosis, which resulted in the administration of a long specific chemotherapy and had negative effects on the patients general condition. In addition, hepatotoxicity and other side effects has been often noted, hand in hand with increased respiratory failure, etc.
14 patients (16%) were diagnosed wrongly with idiopathic fibrosing alveolitis. They received corticosteroid treatment for 6 months (on average). That caused pronounced adverse effects such as hyperglycemia, Cushing's syndrome, hypertension, etc. 7 patients (8%) received, in addition, the immunosuppressive treatment.
In 83 cases (98% of the patients) the diagnosis of PAP was verified histologically in open, videothoracoscopic, or transbronchial lung biopsy. All photographs are of several patients.
Biopsies were fixed in 10% neutral formalin and mounted in paraffin wax. The sections were stained with hematoxylin and eosin, alcian blue, mucicarmine, Congo red, Schiff reagent (PAS-reaction), and according to van Gieson. We also performed cytological examination of sputum and BAL (hematoxylin and eosin staining, according to Giemsa, PAS-reaction).
Thus, along with clinical, radiological, biochemical studies etc. histological investigation was one of the most important stages of our work on the complex PAP diagnostics.
Microscopic examination (fig. 1) showed that the most pronounced changes were detected in subpleural regions. Alveolar walls remained thin, and signs of inflammation or fibrosis were absent; however, there was the hyperplasia of ?-type alveolocytes. In alveoli and bronchioles there was large - or fine-granular eosinophilic substrate (Fig 1a-d). In some places histiocytes, large foamy macrophages, as well as traces of cell detritus and a birefringent cholesterol crystals were visible too (Fig. 1e). There was a positive PAS-reaction in the form of a purple-red color content in the alveoli. Alcian blue and mucicarmine staining were negative. The affected areas had clear boundaries with the surrounding air lung tissue. Sometimes the accumulation of macrophages and giant multinucleated cells with vacuoles (resp. lipids) in the cytoplasm were determined around them (Fig. 1f). There was a weakly expressed infiltration of the septal interstitial tissue with lymphocytes and histiocytes, while mild to moderate fibrosis of alveolar walls was determined as the disease duration was about 1-2 years.
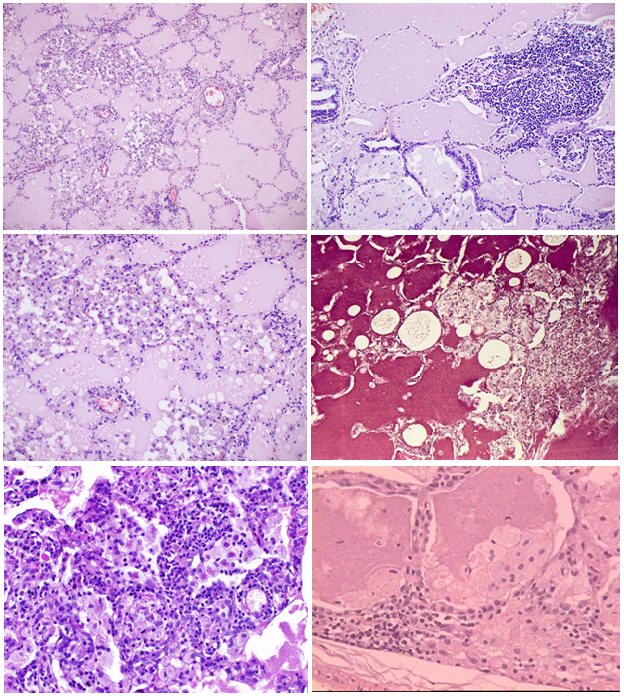
Fig 1: Microscopic picture.
(a) Pale eosinophilic proteinaceous exudate abundantly filling alveolar spaces. Some alveolar walls are infiltraited with lymphoid cells.
(b) Numerous foam macrophages in alveolar spaces, filled with proteinaceous exudate. Slight lymphohistiocytic infiltration of accompanying interalveolar walls.
(c) Massive accumulations of macrophages and lymphoid cells within alveoli. Cell and nuclear debris in some alveoli.
d) Severe lymphohistiocytic infiltration of interstitial tissue as well as mild fibrosis.
(e) Sharply defined rounded holes (lipid droplets) and birefringent cholesterol crystals within alveolar spaces.
(f) Macrophages and multinuclear giant cells around eosinophilic proteinaceous exudate accumulations.
Cytology of sputum and lavage fluid revealed eosinophilic amorphous and granular masses, against which background alveolar macrophages and eosinophilic non-nuclear corpuscles were visible.
Treatments strategy of patients with PAP was determined by the prevalence and severity of lung lesions. As described above2, the only effective method is a total broncho-alveolar lavage under general anesthesia (endotracheal anesthesia).
The following criteria were used as a basic guideline in deciding which type of broncho-alveolar lavage to employ: slowly progressing respiratory failure (hypoxemia during exercise, dyspnea, unproductive cough etc.), chest pain, slightly elevated temperature, fatigue, sweating, weight loss, considerable changes on X-ray with diffuse reticulonodular opacities, thickening of interlobular septa and specific “crazy paving” pattern with “ground glass” opacities in HRCT scans.
On the contrary to the common practice of European authors, we performed usually a single-step lavage procedure of one lung with warm sterile isotonic solution with the addition of N-acetylcysteine. The total quantity of liquid was 1-10 or more liters depending on the volume of lung tissue disturbed. Each patient was performed 1-2, rarely a higher number of procedures.
Thus, after the unilateral total broncho-alveolar lavage improvement according to HRCT was observed in both lungs.
In the case of pronounced respiratory failure segmental broncho-alveolar lavage was performed. The effectiveness of sanation bronchoscopy with such tactics was also quite high, and clinical, functional, and radiological improvement was achieved in 67 patients (79%) whereas over a long period of time remission - in 34 patients (40%).
If necessary, bilateral intervention was implemented and carried out again, but not earlier than 7 days after the first one.
In recent years, segmental broncho-alveolar lavage has been used in our clinic in 9 cases (7 smokers) aged 22-52 (mean age 37,3). This less invasive procedure was performed without general anesthesia and any severe complications. HRCT of PAP and mean functional parameters before and after segmental broncho-alveolar lavage are represented in fig. 2 and table 2.
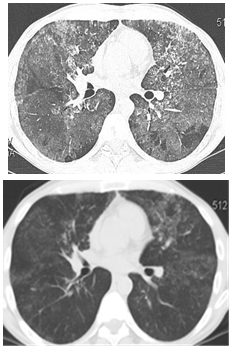
Fig. 2: HRCT of lungs in patient Sh. with type III respiratory failure before (left) and after (right) segmental BAL
Table 2. Functional parameters in patients with PAP (N=9)
| Functional parameters (Mean±SD) | Before multiple segmental lavage | After multiple segmental lavage |
|---|---|---|
| FVC | 71,4±12,7 % | 82±16,0 % |
| FEV1 | 62,7±18,7 % | 69,9±20,0 % |
| DLco | 65,7±15,9% | 81±14,7 % |
| PASP | 34±8 mm Hg | 33±8 mm Hg |
In exceptional cases, showing absence of external breath function disturbances, though there were moderately pronounced changes in X-ray pictures, we came to nothing more than recommending smoking cessation and assignment of N-acetylcysteine, anticipating some chance of the spontaneous remission. There is an evidence that 10-30% of patients with minimal severity of changes in the lungs can show spontaneous remission31, 32. In our patients, the last one was recorded in two cases, when 9 and 12 months after diagnosis of PAP the disappearance of lungs changes was noted in the control HRCT.
Thus, 85 patients were treated with bronchoalveolar lavage. 5-year survival interval was achieved in all patients (100%).
The death causes were secondary infections such as tuberculosis or aspergillosis, as well as the progression of respiratory failure. The prognosis was worsened in case of late diagnosis with long-term unsuccessful antibacterial treatment in particular by tuberculostatics, as well as corticosteroids and physiotherapy, as it has been noted by other authors as well13, 14, 33.
Conclusion
Our observations relate to such a rare lungs pathology, as PAP. They are consistent with the modern ideas about this disease and showed that its characteristic clinical and radiological features give reason for timely diagnosis, despite the fact that etiology of this disease is unknown.
The microscopical examination of the lung biopsy specimens as well as of the sputum smears allowed us to successfully solve the most important diagnostic problems. In fact, within R. Virchow's anatomical thinking it is quite clear that the presence of eosinophilic PAS-positive substrate in alveoli and hyperplasia of ?-type alveolocytes are to a certain extent specific microscopic picture, giving the opportunity properly conduct a differential diagnosis (at an early stage of the disease) of such pulmonary pathology as interstitial lung disease and, first and foremost, idiopathic pulmonary fibrosis (s. usual interstitial pneumonia), tuberculosis, sarcoidosis, pneumocystosis, and some others. This is due to the fact that morphological changes, in essence, go ahead of the clinical manifestations of a particular disease, as has long been suggested by D. S. Sarkisov34, A. I. Strukov35 and other Russian investigators, and is supported recently.
Nevertheless, our own experience shows that the presence of PAS-positive material in the alveoli and bronchioles does not permit to diagnose PAP once and for all. As a matter of fact, they occur more or less frequently in many other lung diseases mentioned above while discussing the problems of PAP differential diagnosis. Subsequently, the diagnostics of PAP needs to take into account the whole complexity of the clinico-radiological, biochemical, immunological, bacteriological, and epidemiological data. It is traditionally the fundamental rule of clinical diagnostics in Russia.
According to our observations, a noticeable improvement was achieved in 67 patients (79%). A set of conditions that are necessary and sufficient for such a result are early and timely diagnosis and a treatment by total or segmental broncho-alveolar lavage. No serious complications were observed. After the timely diagnosis treatment of alveolar proteinosis was uneventful, as evidenced by the 5-year survival rate reaching 100%. Similar results were obtained by Seymour and Presneill7 in the treatment of over 200 patients.
Our further research concerns the investigation of the PAP pathogenesis. Despite a wide range of different factors in 90% of patients with proteinosis is there indeed impaired GM-CSF bioavailability due to anti-GM-CSF autoimmunity, as A.Kumar et al.1 believe?
Acknowledgement
We thank Alexei Mikhailov, MD, Ph.D. Department of Pathology Wake Forest University, Winston-Salem, NC, USA, for his expert assistance with manuscript preparation.
References
-
- Kumar A, Abdelmalak B, Inoue Y, et al. Pulmonary alveolar proteinosis in adults: pathophysiology and clinical approach. Lancet Respir Med. 2018; 6(7): 554-65.
- Ilkovich YM, Ariel BM, Novikova LN, et al. Pulmonary alveolar proteinosis: a long way to correct diagnosis. Problems of diagnostics and therapy routine practice. Annals Clinical & Laboratory Science. 2014; 44 (4): 405-7.
- Danilevskaya O, Averyanov A, Lesnyak V, et al. Confocal laser endomicroscopy for diagnosis and monitoring of pulmonary alveolar proteinosis. J Bronchology Interv Pulmonol. 2015; 22(1): 33-40.
- Ben-Dov I, Segel M. Autoimmune pulmonary alveolar proteinosis: clinical course and diagnostic criteria. Autoimmun Rev. 2014; 13: 513–7.
- Rosen SH, Castleman B, Liebow A. Pulmonary alveolar proteinosis. N Engl J Med. 1958; 258: 1123–42.
- Wang T, Lazar CA, Fishbein MC, et al. Pulmonary alveolar proteinosis. Semin Respir Crit Care Med. 2012; 33(5): 498-508.
- Seymour JF, Presneill JJ. ?ulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med. 2002; 166: 215–35.
- Sawai T, Umeyama Y, Yoshioka S, et al. Autoimmune pulmonary alveolar proteinosis co-existing with breast cancer: a case report. J Med Case Rep. 2014; 8: 279–279.
- Lakshminarayan S, Schwarz MI, Stanford RE. Unsuspected pulmonary alveolar proteinosis complicating acute myelogenous leukemia. Chest. 1976; 69: 433-5.
- Cordonnier C, Fluery-Feith J, Escudier E, et al. Secondary alveolar proteinosis is a reversible cause of respiratory failure in leukemic patients. Am J Resp Crit Care Med. 1994; 149: 788-794.
- Ito K, Iwabe K, Okai T, et al. Rapidly progressive pulmonary alveolar proteinosis in a patient with chronic myelogenous leukemia. Intern Med. 1994; 33(11): 710-3.
- Chaulagain CP, Pilichowska M, Brinckerhoff L, et al. Secondary pulmonary alveolar proteinosis in hematologic malignancies. Hematol Oncol Stem Cell Ther. 2014; 7(4): 127–135.
- Pascual J, Gómez Aguinaga MA, Vidal R, et al. Alveolar proteinosis and nocardiosis in a patient treated by bronchopulmonary lavage. Postgrad Med J. 1989; 65(767): 674–7.
- Sunderland WA, Campbell RA, Edwards MJ. Pulmonary alveolar proteinosis and pulmonary cryptococcosis in an adolescent boy. J Pediatr. 1972; 80(3): 450–6.
- Sauni R, Järvenpää R, Iivonen E, et al. Pulmonary alveolar proteinosis induced by silica dust. Occup Med (Lond). 2007; 57(3): 221–4.
- Keller CA, Frost A, Cagle PT, et al. Pulmonary alveolar proteinosis in a painter with elevated pulmonary concentrations of titanium. Chest. 1995; 108(1): 277–80.
- Likhachev I, Bazura I, Direev VI. The role of several occupational factors in the development of pulmonary alveolar proteinosis (an experimental study). ??? ???. 1975; 37: 63-9. (In Russian).
- Sakai Y, Abo W, Yoshimura H, et al. ?ulmonary alveolar proteinosis in infants. Eur J Pediatr. 1999; 158: 424-6.
- Shah PL, Hansell D, Lawson PR, et al. Pulmonary alveolar proteinosis: clinical aspects and current concepts on pathogenesis. Thorax. 2000; 55(1): 67–77.
- Jouneau S, Kerjouan M, Briens E, et al. Pulmonary alveolar proteinosis. Rev Mal Respir. 2014; 31(10): 975–91.
- Trapnell BC, Uchida K. Pulmonary alveolar proteinosis. Eur Respir Mon. 2009; 46: 208–24.
- Chou CW, Lin FC, Tung SM, et al. Diagnosis of pulmonary alveolar proteinosis: usefulness of papanicolaou-stained smears of bronchoalveolar lavage fluid. Arch Intern Med. 2001; 161(4): 562-6.
- Dail and Hammar´s pulmonary pathology. 3th ed. – Ed. J.F.Tomashefsky (ed.). NY. 2008.
- Khan A, Agarwal R, Aggarwal AN. Effectiveness of granulocyte-macrophage colony-stimulating factor therapy in autoimmune pulmonary alveolar proteinosis: a meta-analysis of observational studies. Chest. 2012; 141 (5): 1273-83.
- Rodríguez Portal JA. Tratamiento de la proteinosis alveolar primaria del adulto. Arch Bronconeumol. 2015; 51(7): 344–9.
- Patel NM, Diaz-Mendoza J, Valdiviezo EA, et al. Case-series of pulmonary alveolar proteinosis treated with bilateral simultaneous whole lung lavage: a novel treatment modality. Am J Respir Crit Care Med. 2015; 191: A4433–A4433.
- Tazawa R, Trapnell BC, Inoue Y, et al. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am J Respir Crit Care Med. 2010; 181: 1345-1354.
- Kavuru MS, Malur A, Marshall I, et al. An open-label trial of rituximab therapy in pulmonary alveolar proteinosis. Eur Respir J. 2011; 38: 1361-67.
- Luisetti M, Rodi G, Perotti C. Plasmapheresis for treatment of pulmonary alveolar proteinosis. Eur Respir J. 2009; 33: 1220-22.
- Tazawa R, Inoue Y, Arai T, et al. Duration of benefit in patients with autoimmune pulmonary alveolar proteinosis after inhaled granulocyte-macrophage colony-stimulating factor therapy. Chest. 2014; 145: 729–37.
- Goldstein LS, Kavuru MS, Curtis-McCarthy P, et al. Pulmonary alveolar proteinosis: clinical features and outcomes. Chest. 1998; 114: 357-62.
- Souza RC, Kanaan D, Martins HP, et al. Spontaneous regression of pulmonary alveolar proteinosis: a case report. Radiol Bras. 2012; 45(5): 294–6.
- Athayde RAB, Arimura FE, Kairalla RA, et al. Characterization and outcomes of pulmonary alveolar proteinosis in Brazil: a case series. J Bras Pneumol. 2018; 44(3): 231-236.
- Sarkisov DS. Are there so-called functional diseases? Clinicheskaya Meditsina. 1994; 2: 71–4. (in Russian).
- Strukov AI. Structure and function in biology and medicine. Future of the science. Etingoff EB (ed.). ?oscow.1978 (in Russian).