
The C-terminal domain of caveolin-1 and pulmonary arterial hypertension: An emerging relationship
Sarah Plucinsky1, and Kerney J. Glover1*
1Department of chemistry, Lehigh university, 6 E. Packer ave., Bethlehem, Pennsylvania, 18015, USA
Abstract
Pulmonary Arterial Hypertension (PAH) is a rare disease that affects the vasculature in the lungs. Currently, there is no cure for PAH, and there appears to be no clear causal factors for the disease. Recently, through whole exome sequencing, caveolin-1, a critical component of cell surface invaginations called caveolae, has been identified as a key protein in the progression of PAH. Specifically, the mutations associated with PAH have been localized to the C-terminal domain of the protein. Since it is known that the C-terminus of caveolin-1 directly interacts with endothelial nitric oxide synthase (eNOS), the link between caveolin-1 and PAH may reside in disrupted nitric oxide (NO) levels, which ultimately triggers the disease state. Recent biophysical studies have now allowed for this relationship to be viewed in a structural context. In this mini-review, we will put the recent structural insights into the C-terminal domain of caveolin-1 into the context of PAH disease progression.
Introduction
Pulmonary Hypertension (PH) is a progressive degenerative disease that affects the vasculature in the lungs and heart. There are several classes of PH: Group 1 pulmonary arterial hypertension (PAH), group 2 PH secondary to left heart disease, group 3 PH secondary to lung disease, group 4 PH associated with chronic thrombo-embolism, and group 5 PH that is associated with several diseases such as renal failure and thyroid disease1. PAH is unique from the other classes because it is not a comorbidity, or in other words, it is not a secondary cause and was initially classified as primary PH2. PAH is characterized by constriction of the vasculature in the lungs that causes an increase in pressure in the pulmonary artery which is responsible for the movement of blood from the heart to the lungs. This buildup of pressure causes immense strain on the heart, and ultimately leads to right ventricular heart failure3. Untreated, the prognosis for patients with PAH is a few years, giving this disease a high morbidity and mortality. While there are several treatments available that can help to alleviate some of the symptoms of PAH, there is no means of reversing or curing the chronic rare disease. Recent studies have improved the genetic understanding of the disease, and these insights could lead to better therapeutic interventions. This is particularly true in the cases of idiopathic PAH and heritable PAH where there is no known outlying cause of the disease. This strongly suggests that there must be a genetic factor that is causing patients to develop PAH4. Studies have shown that mutations to the bone morphogenetic protein receptor type 2 (BMPR2) gene are found in a large percentage of patients that have heritable PAH, but these mutations are also found in cases where there is no familial history of the disease, making the link between BMPR2 and PAH unclear5. Another gene that has been recently identified in the progression of PAH is caveolin-16. The knockout of caveolin-1 leads to several disease-like phenotypes due to the loss of caveolae on the cell surface. In particular, the absence of caveolin-1 leads to vascular deformities and PAH-like symptoms7-10. Furthermore, there is reduced expression of caveolin-1 in the endothelial cells of PAH patients. It has been shown that BMPR2 localizes to caveolae and interacts directly with caveolin-1, and the stabilization of this interaction by the protein elafin has been shown to increase BMPR2 function11,12. Another interaction that is affected by the loss of caveolin-1 is that between endothelial nitric oxide synthase (eNOS) and caveolin-1, which has been shown to have detrimental effects on endothelial health13.
Recently, mutations to the caveolin-1 C-terminal domain have been found in PAH patients6. In this mini-review we present research into the emerging relationship between caveolin-1 and PAH, and how mutations to the caveolin-1 C-terminal domain affect caveolin-1 function, particularly the effects on the caveolin-1/eNOS interaction (Figure 1).
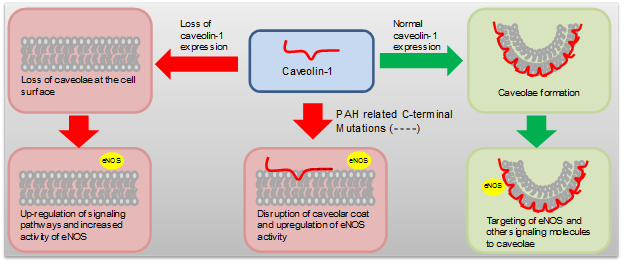
Figure 1: Schematic drawing of the relationship between caveolin-1 expression and PAH
Caveolin-1
Caveolin-1 is a 178 amino acid integral membrane protein that is the primary protein component of 50-100 nm plasma membrane invaginations called caveolae. Caveolin-1 is often represented by four distinct structural domains: a soluble N-terminal domain, a scaffolding domain, a membrane domain, and a C-terminal domain14-17. Recently, secondary structure assignments have shown that the C-terminal domain forms a long single alpha-helix from residues 132-17515. The helix is amphipathic, and is thought to rest on the surface of the membrane14,15. This interfacial interaction with the cell membrane is proposed to be enhanced by three cysteine residues, located in the C-terminal domain, that have been shown to be palmitoylated in vivo. However, the specific role(s) that palmitoylation play(s) in caveolin-1 function is currently unresolved, although it is clear that palmitoylation is not necessary for the proper trafficking of caveolin-1 to caveolae18. The C-terminal domain has also been cited as a key domain for the formation of the higher order oligomer/oligomer interactions that are thought to be critical for stabilizing the highly curved shape of caveolae19. In addition, the C-terminal domain is important for caveolin-1’s interaction with a number of important cellular proteins including connexin and Retrovirus NSP420,21. Recent in vivo studies in cells overexpressing caveolin-1 showed that the fate of the wild-type protein was highly dependent on the tag that was appended to the C-terminus22. This indicates that an unadulterated C-terminal domain may be vital for the proper function of caveolin-1, although more work is needed to completely justify this conclusion. Despite these clear indications that the C-terminal domain is important for caveolin-1 function, little is known about how variations in the C-terminal domain affect caveolin-1 structure and hence cellular behavior.
Caveolin-1 mutations associated with PAH
A recent study of patients with heritable and idiopathic PAH identified a novel frameshift mutation in the caveolin-1 protein, one that occurred in a family with heritable PAH, and the other in a child with idiopathic PAH. This frame shift occurs in the latter portion of the C-terminal domain of caveolin-1 at residue P158 which is identified as P158P fSX22 in the heritable form of the PAH, and P158H fx22 in the patient with idiopathic PAH6. Importantly, there was no evidence of either mutation in control subjects, indicating that the mutation was most likely related to PAH23. Patients experiencing this frameshift mutation showed a marked decrease in endothelial caveolin-1 in their lung tissue6. In addition, studies conducted on the fibroblasts of patients containing the P158P fSX22 mutation revealed that the mutated form of caveolin-1 can still form caveolae, but that the number of caveolae on the cell surface is significantly reduced24. It is interesting to note that both of these mutations occur after the three palmitoylation sites in the caveolin-1 C-terminal domain; therefore, it is unlikely that the reduction in the number of caveolae is due to alterations in caveolin-1 palmitoylation.
A deletion mutation, identified as F160X, has been found in a patient with PAH and congenital generalized lipodystrophy25. This truncated construct lacks the final 20 amino acids in the C-terminal domain, and is similar to the frameshift mutation in that it affects the latter half of the C-terminal domain. Han et al explored the effects of the F160X mutation on the formation and stability of caveolae. While it was found that the mutant protein was able to interact with wild-type caveolin-1, and form caveolae on its own, its oligomeric structure exhibited decreased stability25. In vivo, the C-terminal domain is resistant to immunostaining by C-terminal antibodies. However, when the F160X mutation is introduced, the C-terminal domain is exposed, and able to interact with these antibodies25. These studies show that behavior of the oligomeric complex formed by caveolin-1 is affected by the presence of the C-terminal truncation.
Studies have identified the C-terminal domain as being crucial for the formation of caveolin-1 oligomers19. The study by Han et al supports this by showing that the deletion of the C-terminal residues leads to the formation of less stable oligomers, but does not completely ablate oligomerization. In all of the mutations related to PAH, the last 20 amino acids are affected (either deleted or scrambled). The loss of these amino acids or disruptions, likely affects the amphipathic nature of the C-terminal helix. Consequently, this could affect not only homo-oligomeric interactions, but also lipid/protein interactions as the C-terminal domain has been shown to be important for membrane trafficking and attachment26. The loss of the amphipathic nature due to these mutations may account for a decrease in membrane trafficking, and account for lower levels of caveolin-1 found in PAH patients. This suggests that the amphipathic character of the C-terminal domain may be essential for proper caveolin-1 function.
Caveolin-1, eNOS synthase and PAH
Caveolae are highly abundant, and hence caveolin-1 is highly expressed in endothelial cells. Therefore, caveolin-1 is intimately related to endothelial health. Caveolin-1 null mice have been shown to be viable, but have severe cardiac and vascular defects. These defects are linked to the loss of caveolae on the cell surface8. In PH induced rat model systems, there is a decrease in caveolin-1 expression in endothelial cells, and the upregulation of several cellular pathways before the onset of PH27. In particular, the generation of nitric oxide (NO) by eNOS is affected by caveolin-1 depletion. eNOS is responsible for the production of NO in the vascular system that serves several roles such as the inhibition of platelet aggregation and adhesion28. Caveolin-1 has been shown to directly bind to eNOS, and this interaction inhibits the production of NO within endothelial cells29. Zhou et al studied the progression of PH in caveolin-1 and caveolin-1 and eNOS null mice. These studies revealed that in caveolin-1 null mice there was an increase in eNOS activity that led to PAH, while in the double knockout study (both caveolin-1 and eNOS null mice) there was no PH related vascular defects30. These studies show a direct link between the interaction of caveolin-1 with eNOS and the progression of PAH. It is important to note that there is a high similarity between human caveolin-1 and that of rat and mouse making the aforementioned studies particularly relevant to the human disease (95% and 96% similarity in rat and mouse respectively). In vitro studies monitoring the production of nitrite have shown that the scaffolding and C-terminal domains inhibit eNOS to the same extent as the wild-type protein29. This raises interesting questions about the location of these domains in the structure of caveolin-1. Are these domains spatially close such that they interact simultaneously, or do the two regions interact with eNOS independently? While there are several studies on the interaction of the caveolin-1 scaffolding domain and eNOS, there is a dearth of knowledge on how changes in the C-terminal domain affect caveolin-1/eNOS interactions29,31-33. With the recent identification of C-terminal mutants within caveolin-1, it is now possible to explore how changes in the C-terminal domain affect eNOS behavior in terms of binding and inhibition of NO production. Studies of this type would also give insights into whether the loss of the eNOS interaction or lower levels of caveolae stability is most responsible for PAH progression.
Conclusion
Overall, the role of the caveolin-1 C-terminal domain has been undervalued when studying the cellular function of caveolin-1. With recent structural data, it is now possible to contextualize how mutations to the C-terminal structure may affect caveolin-1 and its interacting partners. Importantly, the caveolin-1 C-terminal domain mutations that are identified in patients with PAH show that this domain is critical for the stability of oligomers that are the bedrock of caveolae homeostasis. Interestingly, the C-terminal domain has also been shown to inhibit eNOS. This interaction is vital for proper vascular health, and disruption of this interaction via mutations in the C-terminal domain could clearly trigger a condition such as PAH. Further research into the role that the C-terminal domain plays in caveolin-1 biology, such as its role in other protein-protein interactions (i.e. BMPR2) will undoubtedly generate new insights into the underlying causal factors of PAH progression. This understanding is critical as there is currently no method for detecting the early stages of PAH within patients; often the disease has progressed to an irreparable extent when it is finally discovered. Promisingly, recent studies have shown that caveolin-1 can be used as a biomarker to help diagnose PAH early by assessing the levels of caveolin-1 expression in endothelial cells34. As more information becomes available on the relationship between caveolin-1 and PAH, it is likely that this new knowledge will facilitate the development of successful therapeutic interventions to mediate the effects of this debilitating disease.
References
- Mathew R. Pathogenesis of Pulmonary Hypertension: A Case for Caveolin-1 and Cell Membrane Integrity. Am J Physiol Heart Circ Physiol. 2013; 306: H15-25.
- Farber HW, Loscalzo J. Pulmonary Arterial Hypertension. N Engl J Med. 2004 351: 1655-1665.
- Thenappan T, Duprez D. Pulmonary Arterial Hypertension. Arterial Disease. 2015; pp 313-332.
- Soubrier F, Chung WK, Machado R, et al. Genetics and Genomics of Pulmonary Arterial Hypertension. J Am Coll Cardiol. 2013; 62: D13-D21.
- The International PC, Lane KB, Machado RD, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta] receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000; 26: 81-84.
- Austin ED, Ma L, LeDuc C, et al. Whole Exome Sequencing to Identify a Novel Gene (Caveolin-1) Associated With Human Pulmonary Arterial Hypertension. Circ Cardiovas Genet. 2012; 5: 336-343.
- Zhao Y, Liu Y, Stan R, et al. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Nat Acad Sci 2002; 99: 11375-11380.
- Drab M, Verkade P, Elger M, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science, 2001; 293: 2449-2452.
- Patel HH, Tsutsumi YM, Head BP, et al. Mechanisms of cardiac protection from ischemia/reperfusion injury: a role for caveolae and caveolin-1. Faseb Journal. 2007; 21: 1565-1574.
- Sehgal PB, Lee JE. Protein trafficking dysfunctions: Role in the pathogenesis of pulmonary arterial hypertension. Pulmonary Circulation. 2011; 1: 17-32.
- Wertz JW, Bauer PM. Caveolin-1 regulates BMPRII localization and signaling in vascular smooth muscle cells. Biochem Biophys Res Commun. 2008; 375: 557-561.
- Nickel NP, Spiekerkoetter E, Gu M, et al. Elafin Reverses Pulmonary Hypertension via Caveolin-1Dependent Bone Morphogenetic Protein Signaling. Am J Respir Crit Care Med. 2015; 191: 1273-1286.
- Razani B, Engelman JA, Wang XB, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001; 276: 38121-38138.
- Parton RG, Hanzal Bayer M, Hancock JF. Biogenesis of caveolae: a structural model for caveolin-induced domain formation. J Cell Sci. 2006; 119: 787-796.
- Plucinsky SM, Glover KJ. Secondary Structure Analysis of a Functional Construct of Caveolin-1 Reveals a Long C-Terminal Helix. Biophys J. 2015; 109: 1686-1688.
- Lee J, Glover KJ. The transmembrane domain of caveolin-1 exibits a helix-break-helix structure. Biochim Biophys Acta. 2012; 1818: 1158-1164.
- Root KT, Plucinsky SM, Glover KJ. Chapter Nine - Recent Progress in the Topology, Structure, and Oligomerization of Caveolin: A Building Block of Caveolae. Curr Top Membr. 2015; 75: 305-336.
- Dietzen DJ, Hastings WR, Lublin DM. Caveolin is palmitoylated on multiple cysteine residues. Palmitoylation is not necessary for localization of caveolin to caveolae. J Biol Chem. 1995; 270: 6838-6842.
- Schlegel A, Lisanti MP. A molecular dissection of caveolin-1 membrane attachment and oligomerization. Two separate regions of the caveolin-1 C-terminal domain mediate membrane binding and oligomer/oligomer interactions in vivo. J Biol Chem. 2000; 275: 21605-21617.
- Mir KD, Parr RD, Schroeder F, et al. Rotavirus NSP4 Interacts with Both the Amino- and Carboxyl- Termini of Caveolin-1. Virus Res. 2007; 126: 106-115.
- Schubert AL, Schubert W, Spray DC, et al. Connexin family members target to lipid raft domains and interact with caveolin-1. Biochemistry. 2002; 41: 5754-5764.
- Han B, Tiwari A, Kenworthy AK. Tagging strategies strongly impact the fate of overexpressed caveolin-1. Traffic. 2015; 16: 417-38.
- Desai AA. Novel Inheritable Caveolin-1 Mutations in Pulmonary Arterial Hypertension. Circ Cardiovasc Genet. 2012; 5: 706-707.
- Marsboom G, Chen Z, Yuan Y, et al. Aberrant Caveolin-1-Mediated Smad Signaling and Proliferation Identified by Analysis of Adenine 474 Deletion Mutation (c.474delA) in Patient Fibroblasts: A New Perspective in the Mechanism of Pulmonary Hypertension. Mol Biol Cell. 2017; E16-06-0380.
- Han B, Copeland CA, Kawano Y, et al. Characterization of a caveolin-1 mutation associated with both pulmonary arterial hypertension and congenital generalized lipodystrophy. Traffic. 2016; 17: 1297-1312.
- Machleidt T, Li WP, Liu P, et al. Multiple domains in caveolin-1 control its intracellular traffic. J Cell Biol. 2000; 148: 17-28.
- HuangJ, Wolk JH, Gewitz MH, et al. Caveolin-1 expression during the progression of pulmonary hypertension. Exp Biol Med. 2012; 237: 956-965.
- Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2011; 33: 829-837.
- Ju H, Zou R, Venema VJ, et al. Direct Interaction of Endothelial Nitric-oxide Synthase and Caveolin-1 Inhibits Synthase Activity. J Biol Chem. 1997; 272: 18522-18525.
- Zhao Y, Zhao YD, Mirza MK, et al. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest. 2009; 119: 2009-2018.
- Trane AE, Pavlov D, Sharma A, et al. Deciphering the binding of caveolin-1 to client protein endothelial nitric-oxide synthase (eNOS): scaffolding subdomain identification, interaction modeling, and biological significance. J Biol Chem. 2014; 289: 13273-13283.
- Bernatchez P, Sharma A, Bauer PM, et al. A noninhibitory mutant of the caveolin-1 scaffolding domain enhances eNOS-derived NO synthesis and vasodilation in mice. J Clin Invest. 2011; 121: 3747-3755.
- Bernatchez PN, Bauer PM, Yu J, et al. Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell-permeable peptides. Proc Nat Acad Sci USA. 2005; 102: 761-766.
- Wang K, Lee M, Ho H, et al. Serum Caveolin-1 as a Novel Biomarker in Idiopathic Pulmonary Artery Hypertension. BioMed Research International. 2015; 2015: 173970.