
A Case report of rare disease Prolidase deficiency in a 15-year-old Pakistan boy
Shahid Ullah1*, Alex Tonks2, Asif Ullah Khan1, Abdulsalam Muharrab Alruwaili3, Muhammad Arif lodhi1
1Abdul Wali khan University, Mardan, KPK, Pakistan
2Division of Cancer and Genetics, Cardiff University School of Medicine, UK
3Northern border university, Saudi Arabia
Abstract
Case presentation
Prolidase enzyme plays a crucial role in proline-rich proteins metabolism and physiological processes such as inflammation, cell proliferation, wound healing, angiogenesis, and carcinogenesis. Due to mutations in the peptidase D (PEPD) gene, the catalytic activity of prolidase loss results in prolidase deficiency. Deficiency of prolidase enzyme is an autosomal inborn metabolic rare genetic disorder that has neither any proper treatment nor consensus for treatment. With approximately 100 cases recorded worldwide, the submitted manuscript describes the 2nd recorded case of prolidase deficiency, an extremely uncommon autosomal recessive disorder associated with collagen metabolism, in a 15-year-old Pakistan boy. The disorder typically becomes apparent during infancy. Affected individuals may have enlargement of the spleen (splenomegaly); in some cases, both the spleen and liver are enlarged (hepatosplenomegaly). Diarrhea, vomiting, and dehydration may also occur. People with prolidase deficiency often develop skin lesions, especially on their hands, feet, lower legs, and face. The severity of the skin involvement, which usually begins during childhood, may range from a mild rash to severe skin ulcers. The severity of symptoms in prolidase deficiency varies greatly among affected individuals. Here we present the report of a 15-year-old boy who has all the clinical manifestations of deficiency of prolidase. This is the 2nd case in Pakistan's 229,488,994 million population.
Introduction
Deficiency of Prolidase enzyme is an autosomal rare recessive genetic disorder caused by mutations in peptidase D (PEPD) gene. This leads to non-healing and recurrent ulcers of the skin and is associated with mental retardation and facial dysmorphism. Mutations in the PEPD gene which encodes prolidase cause prolidase deficiency. This mutation leads to abnormality in amino acid hydroxyproline and proline metabolism1. It is a very rare genetic disorder the worldwide incidence is 1 to 2 cases in one million populations. The first case was reported by Good-men et al. in 1968. Prolidase catabolizes iminodipeptidese containing hydroxyproline and proline. With the deficiency of prolidase deficiency (PD) hydroxyproline and proline increase in urine excretion. Deficiency of proline results in ulcer formation and poor healing of wounds in the body1,2. Prolidase deficiency (PD) is associated with several signs and symptoms. Dermatological manifestations are the most common2. About more than 50% of PD patients have feet and hand ulcers that show high resistance to treatment which results in irregular pattern scarring. Other complications include recurrent chest infections, dysmorphic features, anemia, mental subnormality, most commonly splenomegaly and organomegaly4. Here we present a case report of a 15-year-old boy who has all clinical manifestations of Prolidase deficiency.
Case Report
A 15-year-old boy from Pakistan, born to consanguineous parents, presented with erythematous skin ulcers on the anterior and posterior surface of the left and right legs, buttock, and hand from the age of 4 months. These lesions appeared when the boy was 4 months of age and was extremely painful and associated with the discharge of clear water-like fluid. Until 4 months he was asymptomatic and subsequently developed an ulcer on the right foot as well as on the hand. All possible systemic medicines were prescribed by several clinical physicians, home remedies, including topical treatment with no significant effect (Figure 1) The lesion wound extended and became worse in winter as compared to summer seasons. Over the last several years crusted erythematous plaques appear on both the left and right forearm and both lower legs as well as on the buttock. Previously, the physician reported a case of fungal infection and a dermatologist treated it with different topical medications and antifungals with no aid/relief of symptoms and pain. The boy had normal vital signs 76 beats pulse rate per minute, respiratory rate of 16 /minute, and 110/70 BP. mmHg. He also had deep cutaneous multiple chronic recurrent ulcers on both of his lower legs having raggedy sidelines, the bottom surface is covered by a yellowish color exudate that later becomes dry to form yellowish adherent crusts, which is not fixed to the underline original tissue shown in (Figure 1). There were no varicosities, lymphadenopathy, and pulses were normal. All Other clinical appearances of dermatology comprised, multiple atrophic macules thick dry lesions on the facial region and another part of body telangiectasia on face malar regions, multiple small and linear hyperpigmented atrophic macules on buttocks, and their extensors of extremes dull like telangiectasia of lower legs along with knees, dry fissured soles and palms. Some other manifestations include facial dysmorphism (malaligned teeth, hypertelorism, saddle nose, and high-vaulted palate (Figure 2). But has no single transverse palmar crease, frequent chest, ear, and wound infections, mild mental sub-normality, partial deafness, and short stature as well as splenomegaly. The rest of the systemic examination was unremarkable. His complete blood count report showed hypochromic, microcytic anemia with Hb of 8.1. g/dl, white cell count 15700 /ul with normal platelet, protein in urine ESR is 100 mm/h and hyper IgE >2500 IU/mL. His liver function tests (LFT) and renal function (RFT) were unremarkable as was a chest x-ray. Ultrasound of abdomen showed splenomegaly and mild hepatomegaly. Based on clinical and hematological features including recurrent non-healing ulcer, skin fragility, psychomotometer retardation, short stature, splenomegaly recurrent chest infections microcytic anemia, and characteristics of facial appearance with shallow nasal bridge and hypertelorism, the diagnosis of PD was plausible. No definitive treatment is available for this disorder, but some success is achieved with topical glycine and topical proline reducing the activity of prolidase in dermal fibroblasts leukocytes and erythrocytes, as well as the concentration of proline and hydroxyproline iminodipeptides and recombinant prolidase, is increased urinary excretion. But in Pakistan, there are no facilities that manage the patient health status.
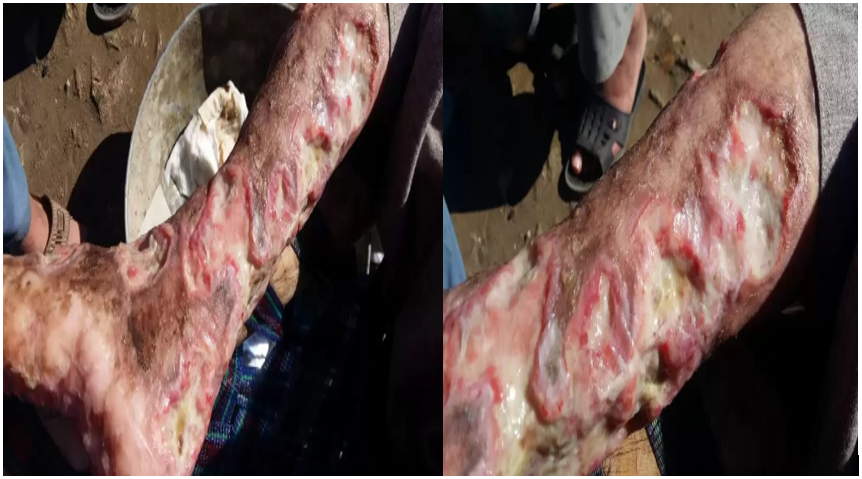
Figure 1: Ulcer on legs

Figure 2: Facial Dysmorphism
Discussion
Goodmen et al. described the first case of PD in 1968. Having mental retardation and non-healing pedal ulcers on both lower bilateral legs5. The gene that is responsible for prolidase is present in chromosome 19q13.11 (GRCh38/hg38), 134 kb spans consist of 15 exons (NM_000285.4) and is transcribed mRNA is 2.3-kb6. PD is a rare autosomal recessive disorder with 1-2 per 1 million individuals. The variant studied in 75 PD patients so far is 35. In which 16 missense/nonsense variants, 9 splice variants, 9 are insertions/deletions and 1 large deletion variants. These variants are distributed along the gene but are almost present on Ct catalytic domain. Nonsense variants do not predict a more severe form of the disease11. Three PEPD Variants are studied recently with the help of whole-exome sequences of the PEPD gene. The Variants c.1409G>A p.(Arg470His) and c.692_694del p.(Tyr231del) were both inherited from the father, While c.575T>C p.(Leu192Pro) was inherited from the mother. The variant p.(Tyr231del) is altering protein dynamics/flexibility and the p.(Leu192Pro) variant causes protein destabilization. The variant p.(Arg470His), has no significant structural differences12. The maternal p.(Leu192Pro) variant and paternal p.(Tyr231del) are together responsible for the proband´s disease. Future whole exome and whole genome studies may help to understand phenotype variability in affected patients and genotype correlations offering clues for future treatments. The hotspot mutations are reported on the 8th, 12th, and 14th exons12. So far,93 cases of prolidase deficiency have been reported in the literature7. The literature about the Pakistani study shows that this will be the 2nd case reported in Pakistan and the first in the male population of Pakistan. The first case was reported in 2017 in a 13-year-old girl1.
Such types of patients have some abnormal facial deformities such as frontal bossing saddle nose, micrognathia, dull expression, mandibular protrusion, and hypertelorism. These patients have a high arched palate, joint laxity, short stature, splenomegaly, mental subnormality, and chronic erosive cystitis.
The most common worse and more critical appearance is the irregular margin ulcers of body skin the most involving the lower legs, and buttock with oozing of clear white fluid. Telangiectasia is most common in PD, crusted lesions on anterior and posterior surfaces of lower legs, face, and hands, soles Dry palms, and have erythematous fissured, and purplish cutaneous lesions3. PD patients have a high prevalence and are more susceptible to infections due to dysfunction of the spleen. In patients with PD, Splenomegaly dysfunction is due to the deposition of amyloid in the spleen8. Iminodipeptides is a degradation product of collagen and the Prolidase enzyme is required to reduce the level of iminodipeptidase.
The decreased level of prolidase results in high dipeptides excretion in urine, that is hydroxyl proline and proline, which is responsible for the abnormal formation of collagen and healing of the wound. PD patient diagnosis can be confirmed by the low level of prolidase in erythrocyte, leukocytes fibroblast, and the high level of proline and hydroxyl proline in urine4. Until now there is no proper and effective treatment for prolidase deficiency. Different systemic and topical treatments are used for the trial and treatment of skin ulcers but the response is still very poor. Different treatments which have been utilized so far have had variable results and included corticosteroid, diphenylhydantoin, apheresis exchange, dapsone, vitamin C (ascorbic acid), and manganese, For activation of prolidase enzyme topical 5 % proline and 5 % glycine blood transfusion containing manganese are used9,10.
Conclusion
PD is an autosomal rare recessive genetic disorder; diagnosis and identification of PD are difficult and even a highly challenging task for treatment. A high competent pediatrician and dermatologist are required for the following evaluation of the affected PD patient to begin early diagnosis and treatment and are very important for family counseling, as well as consanguineous marriage (CM) to prevent the reoccurrences in the affected family.
References
- Khushdil A, Murtaza F. A case of 13-year-old girl with prolidase deficiency. J Ayub Med Coll Abbottabad. 2017; 29(2): 355-357.
- Lopes I, Marques L, Neves E, et al. Prolidase deficiency with hyperimmunoglobulin E: A Case Report. Pediatr Allergy Immunol.2002; 13(2): 140-142. doi:10.1034/j.1399-3038.2002.00075
- Dyne K, Zanaboni G, Bertazzoni M, et al. Mild, lateâonset prolidase deficiency: another Italian case. BrJ Dermatol. 2001; 144(3): 635-636. doi:10.1046/j.1365-2133.2001.04106
- Yaron A, Naider F. Proline-dependent structural and biological properties of peptides and proteins. Crit Rev Biochem Mol Biol.1993; 28(1): 31-81. doi:10.3109/10409239309082572
- Goodman SI, Solomons CC, Muschenheim F, et al. A syndrome resembling lathyrism associated with iminodipeptiduria. Am J Med. 1968; 45(1): 152-159. org/10.1016/0002-9343(68)90016-8
- Monafo V, Marseglia GL, Maghnie M, et al. Transient beneficial effect of GH replacement therapy and topical GH application on skin ulcers in a boy with prolidase deficiency. Pediatr Dermatol. 2000; 17(3): 227-230.
- Kurie BT, D'Sousa A, Bruner BF, et al. Prolidase deficiency breaks tolerance to lupusâassociated antigens. Int JRheum 2013; 16(6): 674-680. doi:10.1046/j.1525-1470.2000.01760
- Bissonnette R, Friedmann D, Giroux JM, et al. Prolidase deficiency: a multisystemic hereditary disorder. J Am Acad Dermatol. 1993; 29(5): 818-821. doi:10.1016/0190-9622(93)70245
- Jemec GB, Moe AT. Topical treatment of skin ulcers in prolidase deficiency. Pediatr Dermatol. 1996; 13(1): 58-60. doi:10.1111/j.1525-1470.1996.tb01191
- Viglio S, Annovazzi L, Conti B, et al. The role of emerging techniques in the investigation of prolidase deficiency: from diagnosis to the development of a possible therapeutical approach. J Chromatogr B Analyt Technol Biomed Life Sci. 2006; 832(1): 1-8. doi:10.1016/j.jchromb.2005.12.049
- Spodenkiewicz M, Spodenkiewicz M, Cleary M, et al. Clinical genetics of prolidase deficiency: an updated review. 2020; 9(5): 108. doi:10.3390/9050108
- Linhares ND, Wilk P, WÄ tor E, et al. Structural analysis of new compound heterozygous variants in PEPD gene identified in a patient with Prolidase Deficiency diagnosed by exome sequencing. Genetics and Molecular Biology. 2021; 44(2): e20200393. doi:10.1590/1678-4685-GMB-2020-0393