
Academic Productivity from Rare Neuromuscular Disease Registries: A Systematic Review
Tran M. Nguyen1, Matt Downs 1, Neil Bennett2, Vitaliy Matyushenko3, Harumasa Nakamura4, Damjan Osredkar5, Shiwen Wu6, Nathalie Goemans7, Anna Ambrosini8, Rahsa El Sherifc9, Craig Campbell1*
1Department of Epidemiology & Biostatistics, Schulich School of Medicine & Dentistry, Western University, London, ON, Canada
2Action Duchenne, London, United Kingdom
3Children with Spinal Muscular Atrophy, Kharkiv, Ukraine
4Translational Medical Center, National Center of Neurology and Psychiatry, Tokyo, Japan
5University Children's Hospital, Ljubljana, Slovenia
6Department of Neurology, the Third medical centre, Chinese PLA (People’s Liberation Army) General Hospital, Beijing, China
7University Hospitals Leuven, Leuven, Belgium
8Fondazione Telethon, Milano, Italy
9Myo-Care Neuromuscular Centre, Cairo, Egypt
Abstract
Background: TREAT-NMD is a global neuromuscular (NM) organization, created to enhance infrastructure to facilitate novel therapeutics reaching patients. One main activity is aimed at supporting NM disease registries. These rare disease registries are useful to fill knowledge gaps for various stakeholders in the disease community using real world data. Although it is important to understand how patient data is being utilized in the TREAT-NMD network and other rare disease registries, there is no systematic process or consistent metric for documenting the academic output from these registries.
Objectives: The objective of this study was to determine the academic output from NM registries associated with the TREAT-NMD network, and the types of research the data is facilitating.
Results: A systematic search of EMBASE, Medline, Cochrane Central and SCOPUS was performed from inception to November 24, 2021. The search yielded a total of 650 results, with 231 full text studies assessed for eligibility and a total of 97 studies that met the inclusion criteria.
Conclusions: The results suggest publications from TREAT-NMD are mainly descriptive or methodologic. Rare disease registries, like the TREAT-NMD network, would benefit from clear and consistent metrics to facilitate reporting of academic output.
Introduction
TREAT-NMD is a neuromuscular network established in 2007 that provides infrastructure to help the development and translation of novel therapies to patients and establish best practice guidelines for neuromuscular disease (NMD) patients worldwide. The network was started as a European Union Network of Excellence, but has since developed into a self-sustaining international organization, uniting patients, clinicians, researchers, and industry in efforts to accelerate therapies for NMD (treat-nmd.org). The organization has several pillars of work with one of the primary efforts to support rare NMD registries. Patient registries have become an important research infrastructure tool for NMD facilitating epidemiologic studies, natural history profiles, clinical trials, and post marketing surveillance. Not only do these patient registries allow eligible patients to be connected to clinical trials, they also allow research and questionnaires on care and disease progression to be efficiently conducted. This is especially important for rare neuromuscular conditions where small patient populations jeopardize clinical trial enrollment. Patient registries serve to mitigate this risk through proper trial planning and recruitment. TREAT-NMD has established an oversight group (TREAT-NMD Global Data-systems Oversight Committee, TGDOC) to support NMD registry development and implementation, sustain relationships, and encourage best practices for NMD registries across the globe. Currently there are over 40 national and international NMD registries focusing on over 10 unique NMDs from over 25 countries affiliated with TGDOC (please see: https://treat-nmd.org/patient-registries/list-of-registries-by-disease for information on each registry). The various NMDs registries include: Charcot marie tooth disease, congenital muscular dystrophies, congenital myasthenic syndromes, Duchenne/Becker muscular dystrophy, Facioscapulohumeral muscular dystrophy, GNE myopathy, Limb girdle muscular dystrophies, MTM and CNM registries, Myotonic dystrophy, and spinal muscular atrophy.
One of the primary objectives of NMD and other rare disease registries are to use the real-world data to fill knowledge gaps for various stakeholders in the disease community. This includes facilitating research and studying the patient populations of interest, with the overall goal of improving patient outcomes. However, there remains sparse literature regarding how to best measure the academic output, such as peer reviewed publications or scientific meeting presentations, from registries and the factors that contribute to productivity. These registries contain vast amounts of patient data and ought to be a source of significant academic activity.
Presently in the TREAT-NMD registry network there is no systematic process of documenting the academic output from the registries, however this is important to understand how patient data is being used in the constituent registries. The purpose of this study is to determine the amount and type academic output from TREAT-NMD registries, and the types of research the data is facilitating.
Materials and Methods
This systematic review followed the PRISMA 2009 guidelines. A systematic search of EMBASE, Medline, Cochrane Central and SCOPUS was performed from inception to November 24, 2021. An additional search of SCOPUS was also performed on November 24, 2021 to identify any relevant studies that mentioned TREAT-NMD in the funding and acknowledgements section. The search strategies are listed in Supplementary Appendix.
Study selection and eligibility criteria
Studies were eligible for inclusion based on two criteria: 1) The primary content of the study was related to a neuromuscular disease and 2) The study finding, or results were generated using data directly from a TREAT-NMD registry. The study must specifically mention that the data was obtained from a TREAT-NMD registry. All study types whether in abstract or published manuscript form were considered for inclusion.
Two authors (CC and RS) independently screened the titles and abstracts, and selected articles for full text review. Full text articles were then reviewed for eligibility, with arbitration by a third author (TMN) for any differences that could not be resolved by consensus. We identified and removed duplicates if the studies appeared in multiple databases and if the same study was presented at multiple academic forums/meetings.
Eligible studies were then sub-grouped into four different categories including: Profile or methodologic, clinical research/guidelines, epidemiologic and basic science. Eligible studies were characterized as either abstracts or published manuscripts.
Results
The search yielded a total of 650 results, with 231 full text studies assessed for eligibility and a total of 97 studies included in the final analysis (Figure 1). The main reason for excluding studies was because they did not include or use TREAT-NMD data in their study.
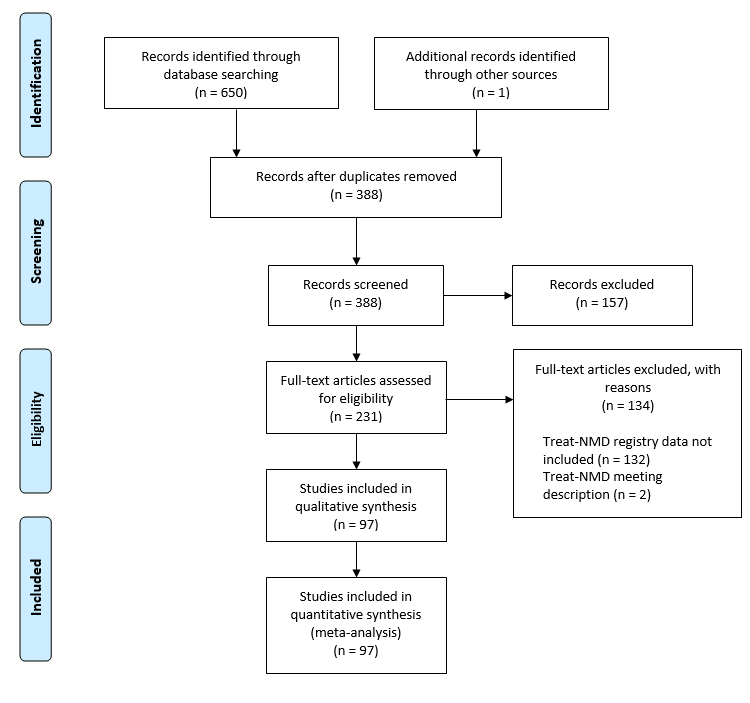
Figure 1: Prisma Diagram
One of the studies was a basic science publication (manuscript), 22 were clinical research/guidelines (seven abstracts and 15 manuscripts) publications, six were epidemiology publications (four abstracts and two manuscripts) and 68 were profile/methodologic publications (31 abstracts and 37 manuscripts). In total, 42 were in abstract form and 55 were published manuscripts (Online Supplement).
It is worthy to note that publications may have been missed if they did not mention TREAT-NMD or TGDOC in the searchable fields, such as the title, abstract, keywords, and acknowledgements.
Discussion
The goal of this systematic review was to assess the type of academic output from the TREAT-NMD Global Database Oversight Committee (TGDOC). The TGDOC is a large network of largely academic and patient organization registries, containing data on thousands of patients1-3. As part of fostering this network, it is important to showcase work from the constituent registries, as well as understanding the different type of academic output from this valuable real-world data. However, it became clear there was limited benchmarking in the literature to fully assess the typical levels of academic productivity from rare disease patient registries. Although rare registries may have different purposes and goals which may not include producing publications, the reality is that there is a substantial amount of patient data in these repositories4. However, the results from this review appear to show that there is a relatively small amount of research addressing epidemiologic or clinical questions that comes directly from these patient repositories. Furthermore, the literature contains little methodological research examining how a registry governance, objectives, and structure link to academic productivity. Therefore, we hope this study contributes to the discussion regarding the need to have consistent metrics and an elevated level of responsibility to report academic productivity that can be anticipated from a rare disease registry.
From this study we can take that many registries do an initial publication highlighting their existence and describing the characteristics and implementation of the registry. This was seen in our sample with 70% of the published/presented academic work was a methodologic description of the registry. There are a smaller number of studies that use the data for addressing a particular scientific question. The clinical studies were largely survey type studies examining patient reported outcomes such as quality of life, burden of illness or adherence to standards of care. Aside from answering relevant scientific questions, publishing from a registry serves many useful purposes including: 1) increasing the awareness of the existence of registry; 2) demonstrating a responsiveness to the patients and NMD disease community; 3) publicizing how the registry is stewarding the data; 4) exposing the registries to constructive criticism on which to drive improvements; 5) contributing to interoperability and collaboration; 6) inviting inquiry for regulatory, medicine authorization and health technology assessment processes; and lastly, 7) strengthening the science around registry methodology5,6.
The primary limitation of our study was that many TREAT-NMD registry studies did not include their affiliation with TREAT-NMD/TGDOC in the searchable fields, such as the title, abstract, and keywords. Thus, the included studies are not an accurate representation of the true number of studies from our constituents. We are aware through internal communications of our member registries of many studies that have used TGDOC data but were not identified in this search for this reason. To overcome this limitation, we subsequently searched SCOPUS database which allowed us to search the funding and acknowledgements section. However, we did not find a significant number of new studies. Going forward, we have appealed to our network registry leaders at our recent annual meeting to include their affiliation with TREAT-NMD in their future publications. Additionally, it is also possible that only the larger NMD centers/registries managed to publish relevant studies from their registries due to larger number of patients included. Smaller registries may need to merge under a TREAT-NMD/TGDOC, in a centralized registry to have the data used in a beneficial way for publication.
TREAT-NMD is a large inclusive organization that aims to advance diagnosis, care and treatment, facilitating the development of novel and existing therapies for patients living with NMD. We believe registries are an integral part of that effort but recognize there are still substantial steps in registry science and utilization efforts that need to occur to inform how registries can be better used as tools to facilitate research. There seems to be a need to develop pathways to better communicate and share registry data with all stakeholders, but more importantly with the patient stakeholders that have offered their valuable data to be included in registries.
Based on this systematic review, we plan to perform an additional similar review to search all rare NMD registries. This will hopefully allow us to achieve the objective of identifying all TREAT-NMD affiliated studies, as well as understand the patterns of academic productivity from registries. With a bigger sample size, we can also begin to try to understand other factors contributing to academic productivity. Factors such as size, location, affiliation with an academic institution, and the prevalence of the disease may influence the publication patterns.
Competing Interest
TMN has no conflicts of interest to declare.
MD has no conflicts of interest to declare.
NB is an employee of TREAT-NMD.
VM has no conflicts of interest to declare.
HN has no conflicts of interest to declare.
DO has no conflicts of interest to declare.
SW has no conflicts of interest to declare.
NG has been clinical trial site investigator for GSK, Biomarin, Elly Lily, Pfizer, Roche, Sarepta, Wave and has served on advisory boards and/or Data Monitoring Committee for Sarepta, Wave, Biomarin, Pfizer, Avidity, Daiichy Sankyo, Avexis, Biogen.
AA has no conflicts of interest to declare.
CC has been a clinical trial site investigator for Acceleron, AMO, Biomarin, Eli Lily, GSK, Biogen, Pfizer, Roche, PTC, Sarepta, Cytokinetics, Wave. Advisory functions: AMO, Biomarin, Acceleron, Biogen, Roche, PTC. DSMB member: Catabasis and Solid.
RES has no conflicts of interest to declare.
References
- Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015; 36(4): 395-402.
- Bladen CL, Thompson R, Jackson JM, et al. Mapping the differences in care for 5,000 spinal muscular atrophy patients, a survey of 24 national registries in North America, Australasia and Europe. J Neurol. 2014; 261(1): 152-163.
- Wood L, Bassez G, Bleyenheuft C, et al. Eight years after an international workshop on myotonic dystrophy patient registries: case study of a global collaboration for a rare disease. Orphanet J Rare Dis. 2018; 13(1): 155.
- Yaneva-Deliverska M. Patient registries for rare diseases. Journal of IMAB - Annual Proceeding (Scientific Papers). 2016; 22: 1166-1168.
- Kodra Y, Weinbach J, Posada-de-la-Paz M, et al. Recommendations for improving the quality of rare disease registries. Int J Environ Res Public Health. 2018; 15(8): E1644.
- (EMA) EMA. Guideline on registry-based studies. 2020.
Supplementary Appendix- Search Strategies
SCOPUS
- Treat-NMD
- Treat-NMD neuromuscular network
- Treat neuromuscular disease
- Treat-neuromuscular disease
- Treat NMD
- TREATNMD
- TGDOC
MEDLINE
- Treat-NMD*.mp.
- Treat-NMD neuromuscular network.mp.
- Treat-neuromuscular disease.mp.
- Treat NMD.mp.
- mp
- mp.
- Or/1-6
- (Becker muscular dystrophy or BMD).mp.
- (Congenital muscular dystrophy or CMD).mp.
- (Congenital myasthenic syndrome or CMS).mp.
- (Charcot marie tooth or CMT).mp.
- (Myotonic dystrophy or DM1 and DM2).mp.
- (Duchenne muscular dystrophy or DMD).mp.
- (Facioscapulohumeral muscular dystrophy or FSHD).mp.
- (GNE myopathy or GNE HIBM).mp.
- (Limb girdle muscular dystrophy or LGMD).mp.
- (Congenital myopathy or Myotubular myopathy or Centronuclear myopathy or CFTD, MTM or CNM or MTM-CNM).mp.
- (Spinal muscular atrophy or SMA).mp.
- (Neuromuscular* or neuromuscular disorder* or neuromuscular disease*).mp.
- Or/8-19
- 7 and 20
EMBASE
- Treat-NMD*.mp.
- Treat-NMD neuromuscular network.mp.
- Treat-neuromuscular disease.mp.
- Treat NMD.mp.
- mp
- mp.
- Or/1-6
- (Becker muscular dystrophy or BMD).mp.
- (Congenital muscular dystrophy or CMD).mp.
- (Congenital myasthenic syndrome or CMS).mp.
- (Charcot marie tooth or CMT).mp.
- (Myotonic dystrophy or DM1 and DM2).mp.
- (Duchenne muscular dystrophy or DMD).mp.
- (Facioscapulohumeral muscular dystrophy or FSHD).mp.
- (GNE myopathy or GNE HIBM).mp.
- (Limb girdle muscular dystrophy or LGMD).mp.
- (Congenital myopathy or Myotubular myopathy or Centronuclear myopathy or CFTD, MTM or CNM or MTM-CNM).mp.
- (Spinal muscular atrophy or SMA).mp.
- (Neuromuscular* or neuromuscular disorder* or neuromuscular disease*).mp.
- Or/8-19
- 7 and 20
Cochrane CENTRAL
#1 Treat-NMD*
#2 Treat-NMD neuromuscular network
#3 Treat-neuromuscular disease
#4 Treat NMD
#5 TreatNMD
#6 TGDOC
#7 #1 or #2 or #3 or #4 or #5 or #6
#8 Becker muscular dystrophy or BMD
#9 Congenital muscular dystrophy or CMD
#10 Congenital myasthenic syndrome or CMS
#11 Charcot marie tooth or CMT
#12 Myotonic dystrophy or DM1 and DM2
#13 Duchenne muscular dystrophy or DMD
#14 Facioscapulohumeral muscular dystrophy or FSHD
#15 GNE myopathy or GNE HIBM
#16 Limb girdle muscular dystrophy or LGMD
#17 Congenital myopathy or Myotubular myopathy or Centronuclear myopathy or CFTD, MTM or CNM or MTM-CNM
#18 Spinal muscular atrophy or SMA
#19 Neuromuscular* or neuromuscular disorder* or neuromuscular disease*
#20 #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19
#21 #7 and #20
Online Supplement
Table 1: Included Studies (N = 97)
|
|
Study |
Study Type |
Publication Type |
|
1 |
Bushby K, Reha A, Northcutt V, et al. First drug registry in Duchenne muscular dystrophy (DMD) to assess Translarna (Ataluren) use, safety, and effectiveness in routine clinical practice. Neuromuscul Disord. 2015;2: S259. |
Abstract |
Clinical Research/Guidelines |
|
2 |
Kirschner J, Rodger S, Vry J, Gramsch K, Lochmuller H, Bushby K. How reference networks develop, implement, and monitor guidelines. Orphanet J Rare. 2012;7: A14. |
Abstract |
Clinical Research/Guidelines |
|
3 |
Mori-Yoshimura M, Mitsuhashi S, Takeuchi F, et al. Clinical research of becker muscular research using nationwide patient registry. Clinical Neurology. 2016;56: S303. |
Abstract |
Clinical Research/Guidelines |
|
4 |
Rodger S, Woods KL, Bladen CL, et al. Care provision for adults with Duchenne muscular dystrophy in the UK: Compliance with international consensus care guidelines. Neuromuscul Disord. 2015;1: S12. |
Abstract |
Clinical Research/Guidelines |
|
5 |
Steffensen B, Otto C, Werlauff U, et al. Health related quality of life in European adults with DMD: Results from the Care-NMD-project. Neuromuscul Disord. October 2015;2: S302. |
Abstract |
Clinical Research/Guidelines |
|
6 |
Dos Santos MR, Goncalves AR, Vieira EM, et al. Molecular profile of 307 Portuguese patients with dystrophinopathy, including 39 new variants. Neuromuscul Disord. 2011;21(9-10):642. |
Abstract |
Clinical Research/Guidelines |
|
7 |
Griggs RC, Herr BE, Reha A, et al. Corticosteroids in Duchenne muscular dystrophy: Major variations in practice. Muscle and Nerve. 2013;48(1):27-31. |
Published manuscript |
Clinical Research/Guidelines |
|
8 |
Landfeldt E, Lindgren P, Bell CF, et al. Compliance to care guidelines for duchenne muscular dystrophy. J Neuromuscul Dis. 2015;2(1):63-72. |
Published manuscript |
Clinical Research/Guidelines |
|
9 |
Landfeldt E, Mayhew A, Straub V, Bushby K, Lochmuller H, Lindgren P. Psychometric properties of the Zarit Caregiver Burden Interview administered to caregivers to patients with Duchenne muscular dystrophy: a Rasch analysis. Disabil Rehabil. 2019;41(8):966-973. |
Published manuscript |
Clinical Research/Guidelines |
|
10 |
Bladen CL, Thompson R, Jackson JM, et al. Mapping the differences in care for 5,000 Spinal Muscular Atrophy patients, a survey of 24 national registries in North America, Australasia and Europe. J Neurol. 2014;261(1):152-163. |
Published manuscript |
Clinical Research/Guidelines |
|
11 |
Landfeldt E, Lindgren P, Bell CF, et al. The burden of Duchenne muscular dystrophy: An international, cross-sectional study. Neuromuscul Disord. 2014;24 (9-10):853. |
Abstract |
Epidemiology |
|
12 |
Lochmuller H, Robertson A, Verhaart IEC, Jones CC, Cook SF. Estimates of spinal muscular atrophy (SMA): Results from the TREAT NMD research program. Neurology Conference: 69th American Academy of Neurology Annual Meeting, AAN. 2017;88(16 Supplement 1) |
Abstract |
Epidemiology |
|
13 |
Vry J, Gramsch K, Rodger S, et al. European cross-sectional survey of current care practices for Duchenne Muscular Dystrophy reveals regional and age-dependent differences. J Neuromuscul Dis. 2016;3(4):517-527. |
Abstract |
Epidemiology |
|
14 |
Koeks Z, Bladen CL, Salgado D, et al. Clinical outcomes in Duchenne Muscular Dystrophy: A study of 5345 patients from the TREAT-NMD DMD global database. J Neuromuscul Dis. 2017;4(4):293-306. |
Published manuscript |
Epidemiology |
|
15 |
Verhaart IEC, Robertson A, Leary R, et al. A multi-source approach to determine SMA incidence and research ready population. J Neurol. 2017;264(7):1465-1473. |
Published manuscript |
Epidemiology |
|
16 |
Aartsma-Rus A, Hoffman E, Bucella F, et al. TREAT-NMD (translational research in Europe, assessment and treatment for neuromuscular disorders). Neuromuscul Disord. October 2015;2: S271. |
Abstract |
Profile/Methodologic |
|
17 |
Bergen Van Den JC, Koeks Z, Straathof CSM, et al. The national Dutch dystrophinopathy patient registry. Neuromuscul Disord. 2013;23 (9-10):775-776. |
Abstract |
Profile/Methodologic |
|
18 |
Bushby K. Advancing diagnosis, care and treatment for people with neuromuscular diseases around the world: A network of excellence to catalyse research infrastructure globally. Orphanet J Rare. 2010;5. |
Abstract |
Profile/Methodologic |
|
19 |
Butoianu N, Sandu C, Iancu D, et al. Preliminary data of national romanian registry of DMD patients. Eur J Paediatr Neurol. 2013;1: S133-134. |
Abstract |
Profile/Methodologic |
|
20 |
Porter B, Cammish P, Turner C, Heslop E, Marini-Bettolo C. The UK myotonic dystrophy patient registry: A key tool in the facilitation of clinical research. J Neuromuscul Dis. 2019;6: S42. |
Abstract |
Profile/Methodologic |
|
21 |
Evangelista T. EURO-NMD, a reference network for neuromuscular diseases. J Neuromuscul Dis. 2019;6 (Supplement 1): S5. |
Abstract |
Profile/Methodologic |
|
22 |
Hammond EL, Youngs L, Bellgard M, Dawkins H. Australasian neuromuscular disease registry. Neuromuscul Disord. 2012;22 (9-10):881. |
Abstract |
Profile/Methodologic |
|
23 |
Hedley V, Bushby K. Promoting rare disease policies across Europe. J Neuromuscul Dis. 2019;6: S6-S7. |
Abstract |
Profile/Methodologic |
|
24 |
Heslop E, Guglieri M, Bushby K, et al. DMD HUB: Expanding clinical trial capacity for Duchenne muscular dystrophy in the UK. Neuromuscul Disord. April 2018;28: S38. |
Abstract |
Profile/Methodologic |
|
25 |
Imber L, Pogoryelova O, Cammish P, et al. GNE myopathy patient registry. J Neuromuscul Dis. 2019;6: S87-S88. |
Abstract |
Profile/Methodologic |
|
26 |
Kimura E, Mori-Yoshimura M, Mitsuhashi S, et al. Current status of dystrophinopathy national registry in Japan. J Neuromuscul Dis. 2016;3: S133-134. |
Abstract |
Profile/Methodologic |
|
27 |
Kimura E, Mori-Yoshimura M, Takahashi PM, et al. Current status of national neuromuscular patient registries in Japan: Remudy. J Neurol Sci. 2017; 381:473. |
Abstract |
Profile/Methodologic |
|
28 |
Kimura E, Nakamura H, Hayashi YK, et al. Infrastructure for new drug development to treat muscular dystrophy - Current status of patient registration in Japan: REMUDY. Neuromuscul Disord. 2012;22 (9-10):882-883. |
Abstract |
Profile/Methodologic |
|
29 |
Kimura E, Nakamura H, Hayashi YK, et al. DMD/BMD patient registry in Japan: Remudy. J Neuromuscul Dis. 2014;1: S127-128. |
Abstract |
Profile/Methodologic |
|
30 |
Oyewole A, Lee J, Leary R, et al. Treat-NMD: Advancing diagnosis, treatment and care in neuromuscular rare diseases. J Neuromuscul Dis. 2018;5: S323-324. |
Abstract |
Profile/Methodologic |
|
31 |
Cammish P, Wood L, Lochmuller H, Gorman G. The UK Myotonic Dystrophy Patient Registry: A key tool in the facilitation of clinical research. Neuromuscul Disord. 2018;28: S17-S18. |
Abstract |
Profile/Methodologic |
|
32 |
Rafferty K, Bowler M, Pohlschmidt M, Rogers M, Turner C, Lochmuller H. New patient registries for Myotonic dystrophy and Facioscapulohumeral muscular dystrophy in the United Kingdom. Neuromuscul Disord. March 2012;1: S34. |
Abstract |
Profile/Methodologic |
|
33 |
Willmann R. The impact of 25 years ENMC workshops. J Neuromuscul Dis. 2018;5: S98. |
Abstract |
Profile/Methodologic |
|
34 |
Karaduman A, Yilmaz O, Alemdarotlu I, Topalotlu H. National registry system for Duchenne Muscular Dystrophy and Spinal Muscular Atrophy. Duchenne muskuler distrofi ve spinal muskuler atrofi hastalari icin ulusal kayit sistemi. Fizyoterapi Rehabilitasyon. 2010;21 (3):226. |
Published manuscript |
Profile/Methodologic |
|
35 |
Karaduman A, Yylmaz O, Alemdaroglu I, Sonmez M, Topaloglu H. Spinal muscular atrophy national registry of Turkey. Neuromuscul Disord. 2010;20 (9-10):671. |
Published manuscript |
Profile/Methodologic |
|
36 |
Nikolenko N, Wood L, Turner C, et al. The UK myotonic dystrophy patient registry. J Neuromuscul Dis. 2016;3: S166-167. |
Published manuscript |
Profile/Methodologic |
|
37 |
Takahashi M, Matsumura T, Takada H, Kuru S, Kimura E. The myotonic dystrophy registry of Japan: Current status and analysis for clinical research. J Neurol Sci. 2017; 381:1078. |
Published manuscript |
Profile/Methodologic |
|
38 |
Wood L, Evangelista T, Norwood F, et al. UK Facioscapulohumeral Muscular Dystrophy (FSHD) patient registry. Orphanet J Rare. 2014;9. |
Published manuscript |
Profile/Methodologic |
|
39 |
Wood L, Evangelista T, Williams M, et al. UK patient registry for facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul Disord. March 2015;1: S36. |
Published manuscript |
Profile/Methodologic |
|
40 |
Xihua L, Lei Z, Chaoping H, Shui zhen Z, Yi W. A comprehensive database of duchenne and becker muscular dystrophy patients in children's hospital of fudan university. J Neurol Sci. 2017; 381:1083-1084. |
Published manuscript |
Profile/Methodologic |
|
41 |
Aartsma-Rus A, Mercuri E, Vroom E, Balabanov P. Meeting report of the "Regulatory Exchange Matters" session at the 5th International TREAT-NMD Conference: Lessons in communication: How an early dialogue between patients, regulators and academics can further therapy development for neuromuscular disorders. Neuromuscul Disord. 2018;28(7):619-623. |
Published manuscript |
Profile/Methodologic |
|
42 |
Ambrosini A, Calabrese D, Avato FM, et al. The Italian neuromuscular registry: A coordinated platform where patient organizations and clinicians collaborate for data collection and multiple usage. Orphanet J Rare. 2018; 13:176. |
Published manuscript |
Profile/Methodologic |
|
43 |
Anonymous. TREAT-NMD A European network for neuromuscular diseases. [German]. TREAT-NMD - Ein europaisches Netzwerk fur neuromuskulare Erkrankungen. Medizinische Genetik. 2009;21(3):375-380. |
Published manuscript |
Profile/Methodologic |
|
44 |
Bladen CL, Rafferty K, Straub V, et al. The TREAT-NMD duchenne muscular dystrophy registries: Conception, design, and utilization by industry and academia. Hum Mutat. 2013;34(11):1449-1457. |
Published manuscript |
Profile/Methodologic |
|
45 |
Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD global database: Analysis of more than 7,000 duchenne muscular dystrophy mutations. Hum Mutat. 2015;36(4):395-402. |
Published manuscript |
Profile/Methodologic |
|
46 |
Bushby K, Lynn S, Straub T, Network T-N. Collaborating to bring new therapies to the patient--the TREAT-NMD model. Acta Myologica. 2009; 28:12-5. |
Published manuscript |
Profile/Methodologic |
|
47 |
Godoy AJ. The creation of a network after an international conference. Neuromuscul Disord. 2011;21 (9-10):722-723. |
Published manuscript |
Profile/Methodologic |
|
48 |
Kimura E, Nakamura H, Mitsuhashi S, et al. Remudy, Japanese national registry for neuromuscular diseases. European Journal of Neurology. June 2016;2):421. |
Published manuscript |
Profile/Methodologic |
|
49 |
Kimura E, Nakamura H, Mitsuhashi S, et al. The infrastructure for the clinical research of muscular dystrophies: Remudy and MDCTN. Clin Neurol. 2014;54(12):1069-1070. |
Published manuscript |
Profile/Methodologic |
|
50 |
Kimura E, Nakamura H, Nishino I. [Remudy]. Review. Brain & Nerve / Shinkei Kenkyu no Shinpo. 2014;66(11):1396-402. |
Published manuscript |
Profile/Methodologic |
|
51 |
Leary R, Oyewole AO, Bushby K, Aartsma-Rus A. Translational Research in Europe for the Assessment and Treatment for Neuromuscular Disorders (TREAT-NMD). Neuropediatrics. 2017;48(4):211-220. |
Published manuscript |
Profile/Methodologic |
|
52 |
Lochmuller H. The TREAT-NMD patient registries for spinal muscular atrophy and Duchenne muscular dystrophy. Dev Med Child Neurol. 2009; 3:6-7. |
Published manuscript |
Profile/Methodologic |
|
53 |
Nakamura H, Kawai M. Registry of muscular dystrophy (Remudy). Construction of the patient self-report registry and collaboration with overseas network. [Japanese]. Clin Neurol. 2011;51(11):901-902. |
Published manuscript |
Profile/Methodologic |
|
54 |
Kimura E, Nakamura H, Hayashi YK, et al. Infrastructure for new drug development to treat muscular dystrophy - Current status of patient registration in Japan: REMUDY. Neuromuscul Disord. 2012;22 (9-10):882-883. |
Published manuscript |
Profile/Methodologic |
|
55 |
Nakamura H, Kimura E, Mori-Yoshimura M, et al. Characteristics of Japanese Duchenne and Becker muscular dystrophy patients in a novel Japanese national registry of muscular dystrophy (Remudy). Orphanet J Rare Dis. 2013; 8:60. |
Published manuscript |
Profile/Methodologic |
|
56 |
Nakamura H, Nishino I, Komaki H, et al. REMUDY-DMD/BMD patient registry in Japan. Neuromuscul Disord. 2010;20 (9-10):670-671. |
Published manuscript |
Profile/Methodologic |
|
57 |
Peay HL, Rangel VM, Brown K, Martin AS, Furlong P. New horizons in the Duchenne Connect registry. Neuromuscul Disord. 2011;21 (9-10):722. |
Published manuscript |
Profile/Methodologic |
|
58 |
Rautenstrauss B, Sereda MW, Walter MC. The prospective German Charcot-Mary-Tooth patient registry. Medizinische Genetik. 2010;22 (1):177. |
Published manuscript |
Profile/Methodologic |
|
59 |
Rodger S, Antonova V, Brabec P, et al. CARE-NMD: The role of patient registries in an international study of care in Duchenne muscular dystrophy. Neuromuscul Disord. 2012;22 (9-10):880. |
Published manuscript |
Profile/Methodologic |
|
60 |
Rodger S, Lochmuller H, Tassoni A, et al. The TREAT-NMD care and trial site registry: An online registry to facilitate clinical research for neuromuscular diseases. Orphanet J Rare Dis. 2013; 8:171. |
Published manuscript |
Profile/Methodologic |
|
61 |
Rodrigues M, Kidd A, Love DR, Roxburgh R. The New Zealand Neuromuscular Disease Registry: Rate of diagnoses confirmed by molecular testing. J Clin Neurosci. 2015;22(2):434-436. |
Published manuscript |
Profile/Methodologic |
|
62 |
Rodrigues MJ, Roxburgh R, Kidd A, et al. The New Zealand neuromuscular disease registry. Neuromuscul Disord. 2011;21 (9-10):723. |
Published manuscript |
Profile/Methodologic |
|
63 |
Takeuchi F, Nakamura H, Mitsuhashi S, et al. National registry of Japanese dystrophinopathy patients: Remudy. Neuromuscul Disord. 2014;24 (9-10):894. |
Published manuscript |
Profile/Methodologic |
|
64 |
Thompson R, Robertson A, Lochmuller H. Natural history, trial readiness and gene discovery: Advances in patient registries for neuromuscular disease. Adv Exp Med Biol. 2017; 1031:97-124. |
Published manuscript |
Profile/Methodologic |
|
65 |
Thompson R, Schoser B, Monckton DG, Blonsky K, Lochmuller H. Patient registries and trial readiness in Myotonic Dystrophy - TREAT-NMD/Marigold international workshop report. Neuromuscul Disord. 2009;19(12):860-866. |
Published manuscript |
Profile/Methodologic |
|
66 |
Wood L, Bassez G, Bleyenheuft C, et al. Eight years after an international workshop on myotonic dystrophy patient registries: Case study of a global collaboration for a rare disease. Orphanet J Rare Dis. 2018; 13:155. |
Published manuscript |
Profile/Methodologic |
|
67 |
Droege M, Finkel RS, Day JW, De Vivo DC, Kirschner J, Mercuri E, et al. The RESTORE registry: A resource for measuring and improving spinal muscular atrophy (SMA) outcomes. Clin Neurol. 2019;59: S402. |
Abstract |
Profile/Methodologic |
|
68 |
Flores D, Ribate MP, Montolio M, Ramos FJ, Gomez M, Garcia CB. Quantifying the economic impact of caregiving for Duchenne muscular dystrophy (DMD) in Spain. Eur J Health Econ. 2020;21(7):1015-23. |
Published manuscript |
Clinical research/guidelines |
|
69 |
Lusakowska A, Kaminska A, Dziala P, Janiszewska K, Grochowski P, Kostera-Pruszczyk A. The role of registry in care and treatment of rare disorders: Polish registry of SMA patients. Neuromuscul Disord. 2019;29: S195-6. |
Abstract |
Profile/Methodologic |
|
70 |
Mellion M, Ronco L, Thompson D, Hage M, Brooks S, van Brummelen E, et al. Phase 1 clinical trial of losmapimod in FSHD: safety, tolerability and target engagement. Neuromuscul Disord. 2019; 29:123. |
Abstract |
Clinical research/guidelines |
|
71 |
Murphy LB, Schreiber-Katz O, Rafferty K, Robertson A, Topf A, Willis TA, et al. Global FKRP Registry: observations in more than 300 patients with Limb Girdle Muscular Dystrophy R9. Ann Clin Transl Neurol. 2020;7(5):757-66. |
Published manuscript |
Clinical research/guidelines |
|
72 |
Nakamura H, Takeda S. Clinical innovation network and status of patient registry for muscular dystrophy (Remudy). Neurotherapeutics. 2019;16 (3):915-6. |
Published manuscript |
Profile/Methodologic |
|
73 |
Schorling DC, Müller CK, Pechmann A, Borell S, Langer T, Thiele S, Walter MC, Zieger B, Kirschner J. Coagulation disorders in Duchenne muscular dystrophy? Results of a registry-based online survey. Acta Myol. 2020;39(1):2-12. |
Published manuscript |
Clinical research/guidelines |
|
74 |
Brabec P, Vondrácek P, Klimes D, Baumeister S, Lochmüller H, Pavlík T, Gregor J. Characterization of the DMD/BMD patient population in Czech Republic and Slovakia using an innovative registry approach. Neuromuscul Disord. 2009;19(4):250-4. |
Published manuscript |
Clinical research/guidelines |
|
75 |
Evangelista T, Wood L, Fernandez-Torron R, Williams M, Smith D, Lunt P, Hudson J, Norwood F, Orrell R, Willis T, Hilton-Jones D, Rafferty K, Guglieri M, Lochmüller H. Design, set-up and utility of the UK facioscapulohumeral muscular dystrophy patient registry. J Neurol. 2016;263(7):1401-8. |
Published manuscript |
Profile/Methodologic |
|
76 |
Jiménez-Moreno AC, Raaphorst J, BabaÄiÄ H, Wood L, van Engelen B, Lochmüller H, Schoser B, Wenninger S. Falls and resulting fractures in Myotonic Dystrophy: Results from a multinational retrospective survey. Neuromuscul Disord. 2018;28(3):229-235. |
Published manuscript |
Clinical research/guidelines |
|
77 |
Landfeldt E, Lindgren P, Bell CF, Guglieri M, Straub V, Lochmüller H, Bushby K. Quantifying the burden of caregiving in Duchenne muscular dystrophy. J Neurol. 2016;263(5):906-915. |
Published manuscript |
Clinical research/guidelines |
|
78 |
Landfeldt E, Lindgren P, Bell CF, Guglieri M, Straub V, Lochmüller H, Bushby K. Health-related quality of life in patients with Duchenne muscular dystrophy: a multinational, cross-sectional study. Dev Med Child Neurol. 2016;58(5):508-15. |
Published manuscript |
Clinical research/guidelines |
|
79 |
Morís G, Wood L, FernáNdez-Torrón R, González Coraspe JA, Turner C, Hilton-Jones D, Norwood F, Willis T, Parton M, Rogers M, Hammans S, Roberts M, Househam E, Williams M, Lochmüller H, Evangelista T. Chronic pain has a strong impact on quality of life in facioscapulohumeral muscular dystrophy. Muscle Nerve. 2018;57(3):380-387. |
Published manuscript |
Clinical research/guidelines |
|
80 |
Pikó H, Vancsó V, Nagy B, Bán Z, Herczegfalvi A, Karcagi V. Dystrophin gene analysis in Hungarian Duchenne/Becker muscular dystrophy families - detection of carrier status in symptomatic and asymptomatic female relatives. Neuromuscul Disord. 2009;19(2):108-12. |
Published manuscript |
Clinical research/guidelines |
|
81 |
Pogoryelova O, Wilson IJ, Mansbach H, Argov Z, Nishino I, Lochmüller H. GNE genotype explains 20% of phenotypic variability in GNE myopathy. Neurol Genet. 2019;5: e308. |
Published manuscript |
Clinical research/guidelines |
|
82 |
Ricci G, Cammish P, Siciliano G, Tupler R, Lochmuller H, Evangelista T. Phenotype may predict the clinical course of facioscapolohumeral muscular dystrophy. Muscle Nerve. 2019;59(6):711-713. |
Published manuscript |
Clinical research/guidelines |
|
83 |
Sárközy A, Bushby K, Béroud C, Lochmüller H. 157th ENMC International Workshop: patient registries for rare, inherited muscular disorders 25-27 January 2008 Naarden, The Netherlands. Neuromuscul Disord. 2008;18(12):997-1001. |
Published manuscript |
Profile/Methodologic |
|
84 |
Bayat F, Sarmiento IG, Ahmadian N, Dehghani Z. "Iranian Registry of Duchenne and Becker Muscular Dystrophies: Characterization and Preliminary Data." J Neuromuscul Dis. 2021; 8(2): 251-259. |
Published manuscript |
Profile/Methodologic |
|
85 |
Das J, Hodgkinson V, Rodrigues M, et al. "SMA -Outcome measures and registries: TREAT-NMD core dataset for SMA - an important tool for post marketing surveillance." Neuromuscul Disord. 2021;31: S129-130. |
Abstract |
Profile/Methodologic |
|
86 |
García-Rodríguez R, Hiller M, Jiménez-Gracia L, et al. "Premature termination codons in the DMD gene cause reduced local mRNA synthesis." Proceedings of the National Academy of Sciences of the United States of America. 2020; 117(28): 15664-16464. |
Published manuscript |
Basic science |
|
87 |
Hodgkinson-Brechenmacher V, McCormick A, Sheriko J, et al. "DMD/BMD -Outcome Measures: A collaborative national Duchenne muscular dystrophy (DMD) registry for real world evidence." Neuromuscul Disord. 2021;31: S85-86. |
Abstract |
Profile/Methodologic |
|
88 |
Imber, L. and V. Straub. "Registries and care of NMD: The international GNE myopathy patient registry." Neuromuscul Disord. 2021; 31: S154-155. |
Abstract |
Profile/Methodologic |
|
89 |
Lowes L, Alfano L, Bendixen R, et al. "Outcome Measures: TREAT-NMD remote outcome measures group - consensus guidelines and recommendations." Neuromuscul Disord. 2021,31: S148. |
Abstract |
Profile/Methodologic |
|
90 |
Lusakowska A, Jedrzejowska M, Kaminska A, et al. "Observation of the natural course of type 3 spinal muscular atrophy: data from the polish registry of spinal muscular atrophy." Orphanet J Rare Dis. 2021; 16:150. |
Abstract |
Epidemiology |
|
91 |
McKenna J, Ambrosini A, Campbell C, et al. "DMD/BMD - Outcome Measures: The TREAT NMD DMD global registry development." Neuromuscul Disord. 2021; 31: S84-S85. |
Abstract |
Profile/Methodologic |
|
92 |
Ogden C, Simon S, McKenna J, et al. "Registries and care of NMD: TREAT-NMD global registries platform." Neuromuscul Disord. 2021; 31: S153-S154. |
Abstract |
Profile/Methodologic |
|
93 |
Porter B, Turner C, Monckton D, et al. "Myotonic Dystrophy: Characterising myotonic dystrophy (DM) and supporting national and international research projects: nine years of the UK DM patient registry." Neuromuscul Disord. 2021; 3: S118-S119. |
Abstract |
Profile/Methodologic |
|
94 |
Raynaud S, Viscidi E, Hall S, et al. Utilization of real-world observational data to study safety and effectiveness of spinal muscular atrophy treatments. Journal of Neuromuscul Disord.2021; 8: S71. |
Abstract |
Profile/Methodologic |
|
95 |
Segovia S, Alfano L, Comi G, et al. "The treat NMD LGMD global registry development." Euro J Neurol.2021; 28: 330. |
Abstract |
Profile/Methodologic |
|
96 |
Sherif RE, Gamal M, Hanafy A. "Registries and care of NMD: The Egyptian neuromuscular registry. 2021; 31: S155. |
Abstract |
Profile/Methodologic |
|
97 |
Turner C, Csimma C, De Luca A, et al. "Registries and care of NMD: Improving and de-risking clinical trials in neuromuscular disease: over 10 years of the TREAT-NMD Advisory Committee for Therapeutics (TACT)." Neuromuscul Disord. 2021;31: S153. |
Abstract |
Profile/Methodologic |