
Can we alter dietary macronutrient compositions and alleviate mitochondrial disease?
W.C. Aw, N. A. Youngson and J. W. O. Ballard*
School of Biotechnology and Biomolecular Sciences, The University of New South Wales, Sydney, Australia
Abstract
Mitochondria are an important regulator of organismal fitness and the key energy converting organelle. The flow of energy in eukaryotes involves the conversion of macronutrients to form substrates that drive mitochondrial respiration. Changing the relative ratio of dietary macronutrients can influence metabolic flexibility and alter the production of mitochondrial metabolites, such as reactive oxygen species (ROS), which can influence mitochondrial functions and affect the organismal health. In this review, we describe the differences in mitochondrial output due to dietary macronutrient composition in individuals with Complex I mutations. Non-synonymous mutations in mitochondrial Complex I subunits are a common cause of early-onset mitochondrial diseases. We discuss the possibility of manipulating macronutrient ratios as a treatment for some cases of mild mitochondrial dysfunction.
Introduction
The influence of diet on mitochondrial functions has been a longstanding question in biomedical science. Discovering the interactions between diet and mitochondria may lead to a tractable mechanism for improving human health and could give an insight into the pathophysiology of mitochondrial diseases1. Mitochondrial functions are determined by a tight coordination between mtDNA, nuclear DNA and the metabolic state of the cells, which is highly influenced by diet. Mechanistically, diet may affect the mitochondrial functions of different mitochondrial haplotypes2,3. Thus, we suggest that it will be necessary to associate the function of mitochondria with diet to provide a more robust understanding of specific mutations in mitochondrial genes.
The subunits of the electron transport chain function to transform the energy contained in nutrients into the high energy bonds of ATP via oxidative phosphorylation (OXPHOS). Energy and substrates that drive mitochondrial respiration can be obtained from either glycolysis of glucose, beta-oxidation of fatty acids or oxidation of amino acids4,5. The end products of mitochondrial respiration differ between these substrate sources, and these differences can exacerbate or relieve mitochondrial disease. In this review, we evaluate the potential for different dietary macronutrient compositions to be used to treat patients with mutations in mitochondrial Complex I.
Macronutrients influence metabolic flexibility
Cells need to adapt fuel oxidation (amino acid oxidation, glycolysis, and beta-oxidation) to fuel availability (carbohydrate, protein and fat) and this is known as “metabolic flexibility” 6. Metabolic flexibility is crucial for an organism to adapt to a variety of physiological conditions by allowing organisms to switch from one type of fuel to another in response to the dietary changes. Regulation of metabolic flexibility is complex and involves compensatory mechanisms including anterograde (nuclear control of mitochondrial functions) and retrograde (nuclear changes in response to mitochondrial signaling) responses7-9. It is possible that metabolic flexibility is primarily substrate driven, as described by the Randle cycle. The Randle cycle is a biochemical mechanism that fine-tunes the balance between glucose and fatty acid oxidation in muscle and adipose tissue. This cycle regulates fuel selection and adapts the supply and demand of the substrate in tissue10.
Changes in dietary macronutrient compositions have recently been suggested to modify the location of electrons entering into the ETC and possibly influence mitochondrial metabolism. For example, electrons that enter through Complex I can generate a total of 10 H+ but only six H+ can be generated when the electrons bypass Complex I and enter through Complex II4 (due to either nutrient availability, diet composition, or complex I mutation). These differences in proton concentration caused by different entry points will influence the transmembrane electrochemical gradient and eventually affect the rate of ATP produced by Complex V2. In the next section, we will discuss the influences of macronutrients on mitochondrial functions. As a model, we will study Complex I mutations to facilitate the understanding of these macronutrient-mitochondrial DNA interactions.
Complex I mutations
Mitochondrial Complex I, or NADH dehydrogenase, is the first entry point of electrons as well as the largest complex in the respiratory chain. It transfers two electrons from NADH to ubiquinone, which simultaneousy facilitates the pumping of four protons across the inner mitochondrial membrane. Mitochondrial Complex I produces approximately 40% of the proton flux that is used in ATP production, and is a major source of ROS production. In worms and flies, knocking down of different Complex I subunits caused increase of lifespan and apparently the relationship is not simply one of reducing ROS resulting in increased longevity11. In human, dysfunctions of mitochondrial Complex I subunits are associated with a range of clinical phenotypes, and these dysfunctions may be caused by mutations in either nuclear or mitochondrial DNA12. The phenotypes of mitochondrial Complex I dysfunctions include reduced fertility, shortened lifespan, progressive neurodegeneration, insulin resistance, type 2 diabetes, mitochondrial encephalomyopathy lactic acidosis and stroke like episodes (MELAS), and Leber heriditary optic neuropathy (LHON)13-21. Given the occurrence of heteroplasmy of mtDNA (wild type and mutant mtDNA) in each cell, the influence of mtDNA mutation can only be detected when the effects of the mutation go beyond a certain threshold that the cell cannot compensate for22.
Influence of dietary protein on Complex I mutations
To date, only a few studies have investigated the relationship between macronutrients and mitochondrial functions. A study on Drosophila has shown that flies fed with high Protein: Carbohydrate (P:C) ratio diet were associated with higher citrate synthase activity which is a biomarker of mitochondrial content3. Additionally, restricted intake of the essential amino acid methionine in rat has been shown to associate with a decrease in mitochondrial content, followed by an increase in mitochondrial functions and decrease in oxidative stress23-25. Indeed, an increased intake of methionine was also shown to increase mitochondrial ROS production and mitochondrial DNA oxidative damage in rat liver26. Combining these findings, it suggests that intake of high protein diet or certain amino acids could increase mitochondrial ROS and mitochondrial content. Amino acids can only generate ATP in a mitochondrial-dependent manner. Thus, more ROS will be produced when proteins are used as the main fuel for energy generation (Figure 1). The increases in ROS lead to increased oxidative stress27,28 which is thought to be a major contributor to the pathogenesis of mitochondrial disease-associated neuronal degradation13,29, reduced fertility30 and diabetes31.
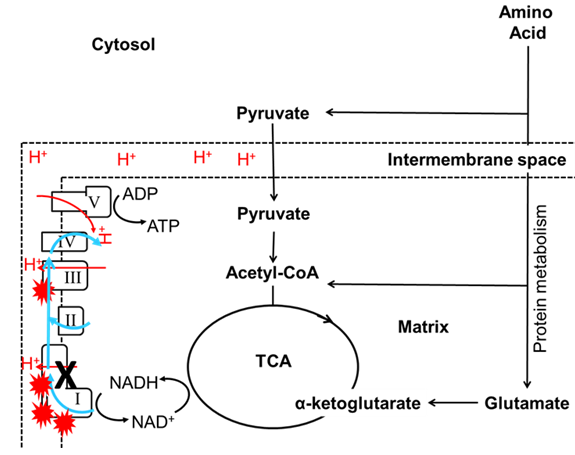
Figure 1: The influence of dietary protein on Complex I mutations. I, II, III, IV and V denoted Complex I, Complex II, Complex III, Complex IV and Complex V respectively. The TCA cycle generates substrates (NADH) for OXPHOS and uses this substrate to fuel mitochondrial respiration. Mitochondrial complexes will collect electrons (blue line) from substrate oxidation and transport protons (red line) into the intermembrane space (dotted line). MtDNA mutations in Complex I (black cross) reduce the efficiency of electron transfer or proton transport, which lead to an increase in ROS production (red explosive symbol). In this case, the amino acids can only generate ATP in a mitochondrial-dependent manner. Thus, more ROS will be produced when proteins are used as the main fuel for energy generation.
Cellular oxygen concentration may also influence ROS formation in a mitotype specific manner. Typically, a lower oxygen content generates less ROS, and a higher content, more ROS32-34. However, there are examples where ROS formation is prevented or at least decreased by increasing the oxygen consumption to decrease cellular oxygen concentration35-37. Likely, mtDNA mutations that cause differences in mitochondrial functions will influence the rates of oxygen consumption38, ATP production39 and therefore ROS formation39,40. Further work on these compounding effects will be important if these interactions are to be fully understood in health and disease.
Simply increasing the amount of mitochondria within a cell to compensate for dysfunction is not always an effective response to mutations in mitochondrial genes. For healthy mitochondria, upregulation of mitochondrial content will cause a small amount of ROS production, which may be beneficial for the organism, because low levels of ROS are required for regulating homeostasis41. However, an increase in mitochondrial content could have a severe impact on organisms that harbour mtDNA mutations28. For example, the His182Tyr substitution in the ND2 (part of Complex I) protein in Drosophila has been shown to increase mitochondrial ROS production and impair energy production3,17. This ND2 mutation induces an increase in mtDNA copy number but with the trade-off of reduced lifespan16,17. Potentially, an upregulation of unhealthy mitochondria will provide a short term benefit for the organism, but may eventually, lead to the accumulation of free radical damage over time and induce cellular cytotoxic stress. In support of this, a recent study in worms has shown that mitochondrial OXPHOS dysfunction will lead to activation of the mitochondrial unfolded protein response, propagation of deleterious mtDNA and potentially mitochondrial diseases42.
Influence of dietary carbohydrate on Complex I mutations
Empirical studies have shown that dysfunction in the mitochondria can cause a switch from mitochondrial respiration to aerobic glycolysis to reduce oxidative stress43-46, which could be considered to be a Warburg-like effect47. In fact, this switch from mitochondrial respiration to aerobic glycolysis was shown to be at least partly mediated by respiratory Complex I48. The missense mutation in mitochondrial ribosomal protein S12 (tko) that reduces Complex I, III and IV activity has been shown to alter the pattern of gene expression in Drosophila49-51. The mitochondrial ribosomal protein S12 is encoded by nuclear genes and responsible for protein synthesis within the mitochondrion. Transcriptome-wide analysis of gene expression shows the tko25tmutant flies experience a metabolism switch in order to compensate the OXPHOS defect. Specifically, the tko25t mutant flies experience an upregulation of lactate dehydrogenase which suggested a switch to glycolysis for ATP production50. Currently, it is not known whether this switch results from Complex I dysfunction, or a retrograde response mediated by ROS.
Here, we suggest that a potential retrograde response for oxidative stress is to switch the energy production from the mitochondrial-dependent (mitochondrial OXPHOS) to the mitochondrial-independent (glycolysis) pathway. We hypothesize that this response can only be initiated in the presence of carbohydrate, thus, diets with high carbohydrate content should provide higher plasticity and have specific advantages for organism that harbour non-synonymous mtDNA mutations in OXPHOS genes. Based on this, we predict that diets rich in sugar could potentially alleviate the mutant phenotype caused by mitochondrial dysfunction. If the hypothesis is supported, it could suggest a potential route for macronutrient management of patients with a subset of mitochondrial diseases. A potential problem with this hypothesis is that the ATP produced from glycolysis would be approximately 20 times lower than that is produced from OXPHOS. However, a switch of energy production from mitochondrial OXPHOS to glycolysis can also lead to lipid accumulation which could force the organism to use lipid as an alternate fuel source46(Figure 2).
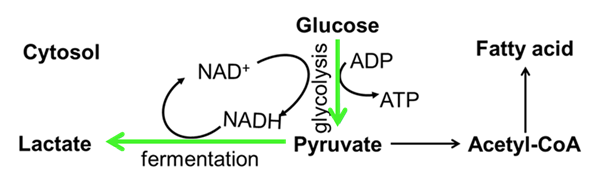
Figure 2: The influence of carbohydrate on Complex I mutations. Mutation in mitochondrial Complex I cause a shift of mitochondrial OXPHOS to glycolysis (green arrow). Hence, less ROS and more lactate are produced. Conversion of pyruvate to lactate will increase NAD+ for continuing the glycolysis process. An accumulation of lactate will eventually lead to the termination of glycolysis. To avoid this, pyruvate will be converted to acetyl-CoA for lipogenesis.
Influence of fat on Complex I mutations
A recent review from Ballard and Youngson2 suggested that changes in dietary macronutrient compositions could modify the entry point of electrons entering into the electron transport system40 and potentially influence mitochondrial functions. It is possible that dietary fat could increase the rate of beta-oxidation and change the concentration of FADH2 and NADH. For example, metabolism of one molecule of glucose produces an NADH: FADH2 ratio of 5:1, whereas long chain fatty acid beta oxidation yields an NADH: FADH2 ratio of about 2:152.
In this case, a slightly deleterious non-synonymous mutation in mitochondrial Complex I may induce more reducing equivalents in the form of fatty acyl-CoA through beta-oxidation of fatty acid and elicit a higher proportion of FADH2. These reduced FADH2cofactors could subsequently shuttle a series of electrons to the ETC through electron transfer flavoprotein (ETF). ETF is a component of ETC that forms a shortcut for transferring electrons from beta-oxidation to ETF-ubiquinone oxidoreductase (ETF-QO)53. The ETF-QO systems could function as a short electron pathway to bypass the slightly deleterious Complex I. Underpinning this hypothesis is evidence showing that the NADH-ubiquinone oxidoreductase 1 alpha subcomplex subunit 5 (NDUFA5) knockout mice have an increase in ETF. This suggests an increase in electron transfer via ETF to UQ may have initiated as a compensatory mechanism to reduce oxidative stress generated from Complex I54.
Which diet is best for Complex I diseases?
There is a world-wide increase in the prevalence of high fat and high carbohydrate diets55. These diets produce high levels of ROS which are major contributors to the development of metabolic diseases56. This environment of hyper nutrition may be exacerbating the phenotypes of Complex I disease patients, potentially pushing individuals with ‘mild/subclinical’ mutations to produce ROS at pathogenic levels. We propose that the studies reported above support the notion that certain diets are more favourable for mitochondrial disease patients with Complex I dysfunction (Table 1).
| Relative ROS production | Relative energy production (ATP) | Relative NADH: FADH2 ratio | |
|---|---|---|---|
| High fat diet (and ketogenic diet) | Low | Sufficient | Low |
| High carbohydrate diet | High i.e. deleterious | Sufficient | High |
| High carbohydrate diet with switch to glycolysis, lipogenesis and beta-oxidation | Low | Sufficient | Low |
| High carbohydrate diet with only glycolysis | Low | Low | High |
| High protein diet | High i.e. deleterious | Sufficient | High |
| High fat and high carbohydrate diet | Extremely high i.e.deleterious | Sufficient | Depends on relative use of beta oxidation, glycolysis and pyruvate in TCA for energy |
Table 1. Predicted variation in the products of diets in Complex I dysfunctionaaDiets that produce low amounts of ROS and sufficient ATP with a low NADH: FADH2 ratio are predicted to be beneficial for individuals with mild OXPHOS Complex I dysfunction.
High protein diets or high carbohydrate diets that produce large amounts of the Complex I donor NADH are unfavourable due to their propensity to produce high amounts of ROS. Reduction of ROS and therefore reduction in oxidative stress could be achieved in two ways. Either through reducing mitochondrial respiration by increasing glycolysis, or bypassing Complex I by increasing the relative amount of beta-oxidation derived fatty acyl-CoA. With increased glycolysis, there is a trade-off between ROS production and energy production so that a glycolysis-reliant metabolism may not produce sufficient energy for good health. However, a shift to beta-oxidation in a high fat diet, or a high carbohydrate diet that has increased lipogenesis, may allow sufficient energy production while minimising ROS production.
Dietary interventions are already among the treatments currently being tested in mitochondrial disease patients. Ketogenic diets (high fat, moderate protein and low carbohydrate) which favour mitochondrial respiration rather than glycolysis for energy metabolism have been tested as therapies for several diseases including cancer, epilepsy and mitochondrial disease57,58. In support of the use of ketogenic diets for patients with Complex I mutations is that the diets increase beta-oxidation (which will produce more fatty acyl-CoA that circumvents Complex I through the ETF-QO systems) and reduce oxidative stress. The diets also increase mitochondrial biogenesis, which could be unfavourable if that increases the amount of mutation-bearing mtDNA. However, studies in cell lines suggest that the diet could actually lead to heteroplasmic shifting which increases the relative amount of non-mutated mtDNA58,59.
Interestingly some of the drug treatments currently being trialed for patients with mitochondrial Complex I dysfunction influence the same processes as the dietary interventions (reviewed in Koopman, et al.60). Drugs such as Curcumin, Idebenone and EPI-743 increase cellular antioxidant levels to reduce ROS levels. Idebenone also delivers electrons directly to complex III, thereby bypassing a deficient Complex I. Coenzyme Q10/ubiquinone supplementation has been tested primarily for its antioxidant properties, but it may also increase the ETF-QO system function to allow bypassing of Complex I.
There are several potential caveats for the therapeutic potential of high fat or high carbohydrate diets. In particular the potential risk of generating obesity in high energy diets. Furthermore, in order to evaluate the potential of the proposed diets for treating patients with Complex I mitochondrial diseases, we also need to determine whether increases in lipogenesis and beta-oxidation are metabolically favourable in high carbohydrate diets. We will also need to determine whether the the ETF-QO system is able to be upregulated sufficiently to meet energy requirements. More work needs to be done in model organisms (e.g. Drosophila, mice) to test the feasibility of these dietary interventions for Complex I diseases. In order to understand the effects of the diets on energy metabolism, measurement of metabolites including ROS, ATP, lactate (as a marker for glycolysis), malonyl-CoA (as a marker for lipogenesis), ketones (as a marker of fat-derived energy metabolism), and the NADH:FADH2 ratio (with fluorescence microscopy)61 would be required. Potentially, the method for measurement of NADH: FADH2 ratio might require some technical development in the living organism.
Conclusions
We propose that rare human diseases that are caused by mutation in mitochondrial Complex I are exacerbated by dietary macronutrient composition. Further, current evidence suggests that diet can modulate mitochondrial functions and affect organismal health. If true, this implies the possibility of manipulating macronutrient ratios as a treatment for some mitochondrial dysfunction-associated diseases.
References
- Wallace DC, Fan W. The pathophysiology of mitochondrial disease as modeled in the mouse. Genes & development. 2009;23(15):1714-36.
- Ballard JWO, Youngson NA. Can diet influence the selective advantage of mitochondrial DNA haplotypes? Bioscience Reports. 2015.
- Pichaud N, Messmer M, Correa CC, Ballard JWO. Diet influences the intake target and mitochondrial functions of Drosophila melanogaster males. Mitochondrion. 2013;13(6):817-22.
- Nelson DL, Lehninger AL, Cox MM. Lehninger principles of biochemistry. W. H. Freeman; 2008.
- Muoio DM. Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell. 2014;159(6):1253-62.
- Meex RC, Schrauwen-Hinderling VB, Moonen-Kornips E, Schaart G, Mensink M, Phielix E, et al. Restoration of muscle mitochondrial function and metabolic flexibility in type 2 diabetes by exercise training is paralleled by increased myocellular fat storage and improved insulin sensitivity. Diabetes. 2010;59(3):572-9.
- Wallace DC. Mitochondria, Bioenergetics, and the Epigenome in Eukaryotic and Human Evolution. Cold Spring Harbor symposia on quantitative biology. 2009;74:383-393.
- Whelan SP, Zuckerbraun BS. Mitochondrial Signaling: Forwards, Backwards, and In Between. Oxidative Medicine and Cellular Longevity. 2013;2013:10.
- Almeida LS, Nogueira C, Vilarinho L. Nuclear-mitochondrial intergenomic communication disorders. 2012.
- Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1(7285):785-9.
- Hur JH, Stork DA, Walker DW. Complex-I-ty in aging. J Bioenerg Biomembr. 2014;46(4):329-35.
- Swalwell H, Kirby DM, Blakely EL, Mitchell A, Salemi R, Sugiana C, et al. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. Eur J Hum Genet. 2011;19(7):769-75.
- Schon EA, Manfredi G. Neuronal degeneration and mitochondrial dysfunction. J Clin Invest. 2003;111(3):303-12.
- Burman JL, Itsara LS, Kayser EB, Suthammarak W, Wang AM, Kaeberlein M, et al. A Drosophila model of mitochondrial disease caused by a complex I mutation that uncouples proton pumping from electron transfer. Dis Model Mech. 2014;7(10):1165-74.
- Aw WC, Correa CC, Clancy DJ, Ballard JWO. Mitochondrial DNA variants in Drosophila melanogaster are expressed at the level of the organismal phenotype. Mitochondrion. 2011;11(5):756-63.
- Clancy DJ. Variation in mitochondrial genotype has substantial lifespan effects which may be modulated by nuclear background. Aging Cell. 2008;7(6):795-804.
- Correa CC, Aw WC, Melvin RG, Pichaud N, Ballard JWO. Mitochondrial DNA variants influence mitochondrial bioenergetics in Drosophila melanogaster. Mitochondrion. 2012;12(4):459-64.
- Selvi Rani D, Vanniarajan A, Gupta NJ, Chakravarty B, Singh L, Thangaraj K. A novel missense mutation C11994T in the mitochondrial ND4 gene as a cause of low sperm motility in the Indian subcontinent. Fertil Steril. 2006;86(6):1783-5.
- van de Weijer T, Sparks LM, Phielix E, Meex RC, van Herpen NA, Hesselink MKC, et al. Relationships between mitochondrial function and metabolic flexibility in type 2 diabetes mellitus. PLoS ONE. 2013;8(2):e51648.
- Kirches E. LHON: Mitochondrial mutations and more. Current Genomics. 2011;12(1):44-54.
- Swalwell H, Kirby DM, Blakely EL, Mitchell A, Salemi R, Sugiana C, et al. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. Eur J Human Genet. 2011;19(7):769-775.
- Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16(9):530-42.
- Sanz A, Caro P, Ayala V, Portero-Otin M, Pamplona R, Barja G. Methionine restriction decreases mitochondrial oxygen radical generation and leak as well as oxidative damage to mitochondrial DNA and proteins. FASEB J. 2006;20(8):1064-73.
- Perrone CE, Mattocks DA, Jarvis-Morar M, Plummer JD, Orentreich N. Methionine restriction effects on mitochondrial biogenesis and aerobic capacity in white adipose tissue, liver, and skeletal muscle of F344 rats. Metabolism. 2010;59(7):1000-11.
- Caro P, Gomez J, Sanchez I, Naudi A, Ayala V, Lopez-Torres M, et al. Forty percent methionine restriction decreases mitochondrial oxygen radical production and leak at complex I during forward electron flow and lowers oxidative damage to proteins and mitochondrial DNA in rat kidney and brain mitochondria. Rejuvenation Res. 2009;12(6):421-34.
- Gomez J, Caro P, Sanchez I, Naudi A, Jove M, Portero-Otin M, et al. Effect of methionine dietary supplementation on mitochondrial oxygen radical generation and oxidative DNA damage in rat liver and heart. J Bioenerg Biomembr. 2009;41(3):309-21.
- Kasahara E, Sekiyama A, Hori M, Hara K, Takahashi N, Konishi M, et al. Mitochondrial density contributes to the immune response of macrophages to lipopolysaccharide via the MAPK pathway. FEBS Lett. 2011;585(14):2263-2268.
- Passos JF, von Zglinicki T, Kirkwood TB. Mitochondria and ageing: winning and losing in the numbers game. Bioessays. 2007;29(9):908-17.
- Carelli V, Rugolo M, Sgarbi G, Ghelli A, Zanna C, Baracca A, et al. Bioenergetics shapes cellular death pathways in Leber's hereditary optic neuropathy: a model of mitochondrial neurodegeneration. Biochim Biophys Acta. 2004;1658(1-2):172-9.
- Venkatesh S, Deecaraman M, Kumar R, Shamsi MB, Dada R. Role of reactive oxygen species in the pathogenesis of mitochondrial DNA (mtDNA) mutations in male infertility. Indian J Med Res. 2009;129(2):127-37.
- Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal. 2010;12(4):537-77.
- Maher P, Schubert D. Signaling by reactive oxygen species in the nervous system. Cell Mol Life Sci. 2000;57(8-9):1287-305.
- Sauer H, Wartenberg M, Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem. 2001;11(4):173-86.
- Hao Y, Cheng D, Ma Y, Zhou W, Wang Y. The relationship between oxygen concentration, reactive oxygen species and the biological characteristics of human bone marrow hematopoietic stem cells. Transplant Proc. 2011;43(7):2755-61.
- Barja G. Mitochondrial oxygen consumption and reactive oxygen species production are independently modulated: implications for aging studies. Rejuvenation Res. 2007;10(2):215-24.
- Turrens JF. Superoxide Production by the Mitochondrial Respiratory Chain. Bioscience Reports. 1997;17(1):3.
- Casteilla L, Rigoulet M, Pénicaud L. Mitochondrial ROS Metabolism: Modulation by Uncoupling Proteins. IUBMB Life. 2001;52(3-5):181-188.
- Bank W, Chance B. Diagnosis of defects in oxidative muscle metabolism by non-invasive tissue oximetry. Mol Cell Biochem. 1997;174(1-2):7-10.
- Vojtíšková A, Ješina P, Kalous M, Kaplanová V, Houšt?k J, Tesa?ová M, et al. Mitochondrial Membrane Potential and ATP Production in Primary Disorders of ATP Synthase. Toxicology Mechanisms and Methods. 2004;14(1-2):7-11.
- Voets AM, Huigsloot M, Lindsey PJ, Leenders AM, Koopman WJ, Willems PH, et al. Transcriptional changes in OXPHOS complex I deficiency are related to anti-oxidant pathways and could explain the disturbed calcium homeostasis. Biochim Biophys Acta. 2012;1822(7):1161-8.
- Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48(2):158-67.
- Lin YF, Schulz AM, Pellegrino MW, Lu Y, Shaham S, Haynes CM. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature. 2016;533(7603):416-9.
- Morris G, Berk M. The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med. 2015;13(1):1-24.
- Jayasena T, Poljak A, Braidy N, Smythe G, Raftery M, Hill M, et al. Upregulation of glycolytic enzymes, mitochondrial dysfunction and increased cytotoxicity in glial cells treated with Alzheimer’s disease plasma. PLoS ONE. 2015;10(3):e0116092.
- O'Donnell J, Ahuja GD. Drug Injury: liability, analysis, and prevention. Lawyers & Judges Publishing Company; 2005.
- Lee S-J, Zhang J, Choi AMK, Kim HP. Mitochondrial dysfunction induces formation of lipid droplets as a generalized response to stress. Oxidative Medicine and Cellular Longevity. 2013;2013:327167.
- Senyilmaz D, Teleman AA. Chicken or the egg: Warburg effect and mitochondrial dysfunction. F1000Prime Reports 2015;7:41
- Calabrese C, Iommarini L, Kurelac I, Calvaruso MA, Capristo M, Lollini P-L, et al. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer & Metabolism. 2013;1(1):1-15.
- Jacobs HT, Fernández-Ayala DJM, Manjiry S, Kemppainen E, Toivonen JM, O'Dell KMC. Mitochondrial disease in flies. Biochimica et Biophysica Acta (BBA) – Bioenergetics. 2004;1659(2–3):190-196.
- Fernández-Ayala DJM, Chen S, Kemppainen E, O'Dell KMC, Jacobs HT. Gene expression in a drosophila model of mitochondrial disease. PLoS ONE 2010;5(1):e8549.
- Chen S, Oliveira MT, Sanz A, Kemppainen E, Fukuoh A, Schlicht B, et al. A cytoplasmic suppressor of a nuclear mutation affecting mitochondrial functions in drosophila. Genetics. 2012;192(2):483-493.
- Salway JG. Metabolism at a Glance. Blackwell Scientific Publications; 1994.
- Watmough NJ, Frerman FE. The electron transfer flavoprotein: Ubiquinone oxidoreductases. Biochimica et Biophysica Acta (BBA) – Bioenergetics. 2010;1797(12):1910-1916.
- Peralta S, Torraco A, Wenz T, Garcia S, Diaz F, Moraes CT. Partial complex I deficiency due to the CNS conditional ablation of Ndufa5 results in a mild chronic encephalopathy but no increase in oxidative damage. Human Molecular Genetics. 2013
- Popkin BM, Adair LS, Ng SW. Global nutrition transition and the pandemic of obesity in developing countries. Nutr Rev. 2012;70(1):3-21.
- Rani V, Deep G, Singh RK, Palle K, Yadav UC. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016;148:183-93.
- Ahola S, Auranen M, Isohanni P, Niemisalo S, Urho N, Buzkova J, et al. Modified Atkins diet induces subacute selective ragged-red-fiber lysis in mitochondrial myopathy patients. EMBO Mol Med. 2016.
- Branco AF, Ferreira A, Simoes RF, Magalhaes-Novais S, Zehowski C, Cope E, et al. Ketogenic diets: from cancer to mitochondrial diseases and beyond. Eur J Clin Invest. 2016;46(3):285-98.
- Santra S, Gilkerson RW, Davidson M, Schon EA. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann Neurol. 2004;56(5):662-9.
- Koopman WJ, Beyrath J, Fung CW, Koene S, Rodenburg RJ, Willems PH, et al. Mitochondrial disorders in children: toward development of small-molecule treatment strategies. EMBO Mol Med. 2016;8(4):311-27.
- Bartolome F, Abramov AY. Measurement of mitochondrial NADH and FAD autofluorescence in live cells. Methods Mol Biol. 2015;1264:263-70.