
Clinical and Genetic Characterization of Cystinosis: Unmet Healthcare Needs in a Cohort Study from a Developing Country
Paola Krall1,2* Jean Grandy3*, Lillian Bolte4,5*, Jennie Salgado6, Felipe Cavagnaro5, Claudia González7,8, Jose Luis Guerrero2,9*
1 Institute of Medicine, Faculty of Medicine, Universidad Austral de Chile, Valdivia.
2 Department of Pediatrics and Child Surgery, Faculty of Medicine, University of Chile, Santiago de Chile.
3 Department of Nephrology, Hospital Exequiel González Cortés, Santiago de Chile.
4 Department of Nephrology, Hospital Roberto del Río, Santiago de Chile.
5 Department of Pediatrics, Clínica Alemana de Santiago, Santiago de Chile.
6 Department of Nephrology, Hospital CQ Herminda Martín, Chillán.
7 Department of Nephrology, Hospital Sótero del Río, Santiago de Chile
8 Department of Pediatrics, Pontificia Universidad Católica de Chile, Santiago de Chile
9Department of Nephrology, Hospital Dr. Luis Calvo Mackenna, Santiago de Chile.
(*) all these authors contributed equally.
Abstract
Background
Cystinosis is a rare disease caused by CTNS gene defects. The main clinical presentations are nephropathic infantile cystinosis (NIC) and nephropathic juvenile cystinosis (NJC); both develop chronic kidney disease (CKD) and extrarenal complications. Opportune diagnosis and access to therapy are challenging in developing countries.
Methods
To describe the clinical and genetic profile in all cystinosis patients known to be diagnosed in Chile, we performed a retrospective review of the medical records of those patients diagnosed from 1994 to 2022. Age at diagnosis, glomerular filtration rate, metabolic variables, anthropometric values, access to treatment, outcomes, and genetic results were analyzed.
Results
Nine patients (8NIC/1NJC) were diagnosed. Patients with NIC had a median age of 16.5 (IQR 13-23) months at diagnosis, and two patients died during follow-up. Most of the patients started cysteamine therapy up to 5 months after diagnosis and reached CKD stages 3-4 within four years. During the follow-up, all but one of the NIC patients showed height for age Z-score values between -1.5 and -4.0. Two patients received kidney transplants, and one of them remains functional after 15 years. The single NJC was a 21-year-old female patient who received irregular cysteamine therapy and rapidly reached CKD stage 5. Genetic testing was positive in 7/7 cases, being del57kb the predominant variant (10/14 alleles).
Conclusions
Developing countries face many challenges in providing adequate healthcare. Our findings show clinical and diagnostic aspects to the medical and patient community that might contribute to the diagnostic approach and treatment access for cystinosis in Chile. Opportune genetic testing may facilitate early diagnosis that is known to be associated with a better prognosis.
Introduction
Cystinosis is an autosomal recessive disease associated with biallelic variants in CTNS (MIM#606272) that encodes the cystinosin protein. This protein acts as a defective lysosomal transporter in patients with cystinosis, causing cystine accumulation throughout multiple organs and their progressive deterioration1,2.
Cystinosis affects 1 in 100,000-200,000 individuals with varying clinical manifestations depending on the disease onset and severity3,4. Nephropathic infantile cystinosis (NIC; OMIM#219800) is the predominant form, presenting symptoms within the first year of life. This clinical form is the most severe and eventually progresses to chronic kidney disease (CKD) during the first decade of life, often requiring kidney replacement therapy (KRT). A kidney transplant (KT) is usually required without evidence of recurrence in allografts. KRT and the involvement of other organs can be delayed with optimal clinical management5,6. Nephropathic juvenile cystinosis (NJC; OMIM#219900) is characterized by late-onset of symptoms, typically during adolescence, and together with NIC cause 95% of all cases. NJC patients require KRT later in life compared to NIC patients.
Traditionally, NIC/NJC is diagnosed by measuring intra-leukocyte cystine, but it is not usually available in developing countries and genetic testing has evolved as a feasible diagnostic tool. Genetic diagnosis yields a mutation detection rate >95% and has revealed a broad spectrum of CTNS variants7. More than 150 variants have been identified, but a 57-kb deletion (del57kb) involving a major portion from CTNS is the most common variant identified in patients from Europe and North America, with a lower frequency in the Middle East8.
Cysteamine is the only available drug to improve renal and extrarenal prognosis9. Opportune diagnosis and treatment with cysteamine are still challenging in some countries since the disease is infrequent. In addition, limited resources related to the health system and/or unequal drug distribution, evidence a significant gap in the clinical evolution of patients from developing and developed nations10,11.
We aimed to describe clinical and genetic determinants, access to cysteamine, and the long-term follow-up in all cystinosis patients known to be diagnosed in Chile.
Methods
We conducted a retrospective review of the clinical records and genetic analysis in 9 cystinosis patients diagnosed in Chile between 1994, when the first medical registry was known, and March 2022. We analyzed characteristics at disease onset to classify each case as NIC or NJC, annual monitoring of biochemical variables [serum creatinine, calcium, phosphorus, plasma bicarbonate, intact parathyroid hormone (PTH), free thyroxine, thyroid-stimulating hormone, proteinuria], recombinant human growth hormone (rhGH) use and anthropometric values according to World Health Organization/Centers for Disease Control and Prevention. To evaluate anthropometric values we used Z-score that represent the distance between a data value and the mean value in a reference population (same age and gender), by using standard deviations. The formula to calculate Z-score is: (Patient data value – mean value in the reference population) / (Standard deviation value in the reference population)12.
In addition, other complications were registered: corneal crystals, exocrine or endocrine dysfunction, myopathies, neurocognitive impairment, swallowing difficulties, and bone mineral disturbances. Conditions at the time of recruitment were recorded as alive or deceased, with or without KT. To determine kidney function in each patient at annual controls, glomerular filtration rate (eGFR) was estimated according to Schwartz's formula while the CKD-EPI equation was used in patients over 18 years old13,14.
Confirmatory diagnosis of cystinosis was made with intra-leukocyte cystine levels measured overseas until 2015 and has been performed by CTNS genetic analysis in Chile since 2016. Regarding cysteamine therapy for each patient, we registered their health system (public vs. private) and their funding sources. We evaluated the time between the NIC/NJC diagnosis and access to the first cysteamine dose. The oral doses were adjusted during follow-up according to established recommendations of body weight or surface area. The use of cysteamine eye drops was also recorded. It was not possible to evaluate therapy adequacy and compliance because intra-leukocyte cystine measurement is not available in Chile. Routine therapy for biochemical abnormalities and CKD complications was provided.
Genetic testing was offered to 7 patients and 9 relatives. Their samples were collected between 2016 and March 2022, all declaring themselves to be Chileans. DNA was isolated using the Whole Blood Genomic DNA Purification kit (ThermoFisher®). Firstly, the specific del57kb PCR was performed in all samples as follows: multiplex PCR containing LDM forward (5´-CTA ACA GTA TCA CCG GAG TC-3´), LDM reverse (5´-GGC CAT GTA GCT CTC ACC TC-3´), D17S829 forward (5´-CTA GGG GAG CGT GTT AGC AT-3´) and D17S829 reverse (5´-TGT AAG ACT GAG GCT GGA GC-3´) primers each one at a final concentration 0.5 μM, as well as a ready-to-use solution (GoTaq® Green Master Mix) and 1ul DNA at ~60 ng/ul was performed. The final products were analyzed by electrophoresis where the presence of the del57kb yielded a 423-bp PCR product, while the D17S829 primers yielded 260-290bp product. This approach is able to discriminate homozygous non-deleted, heterozygous and homozygous deleted genotypes15. Two patients with negative results (homozygous non-deleted) were considered for subsequent analysis by direct sequencing on the CTNS exons 3-12. Sequence analysis was performed using the SeqScape v2.5 software (Applied Biosystems purchased license) to align the obtained sequences with the reference sequence NM_001031681.2 downloaded from the UCSC genome browser (http://genome.ucsc.edu)16. We checked if the detected variants were in databases (HGMD, ClinVar) and analyzed them with predictive bioinformatic tools (Mutation taster, SIFT, and Polyphen-2)17-19. For each variant, segregation analysis was performed in the family by targeted analysis, LDM/D17S829 PCR, or exon sequencing, to establish inheritance and/or to provide genetic counseling if requested. Once a genetic cause was confirmed in the index case, we offered a genetic analysis targeted to the affected alleles to the parents and other at-risk relatives15.
Statistical Analysis
Descriptive statistical analysis methods were used to analyze clinical records and genetic data. Continuous variables were described with median and interquartile ranges [IQR]. Qualitative variables were described with range or percentage. The images describing the evolution of the estimated glomerular filtration rate and height-for-age Z-scores were elaborated on the GraphPad Prism 9 software.
Ethics Concerns
Ethical approval was obtained from the corresponding Institutional Review Board and local Hospital Ethics Committee. This study was performed following the Helsinki Declaration, Good Clinical Practice and Chilean Legislation (Laws nr. 20.120, 20.584 and 19.628). All legal guardians of pediatric patients and adult participants signed an informed written consent agreeing to be included in this study for retrospective data collection and to provide a blood sample for genetic analysis.
Results
Basic patients characteristics
Nine patients with cystinosis were diagnosed between 1994 and March 2022 at Chilean nephrology units. The diagnosis of 8 cases was NIC at a median age of 16.5 (IQR, 13-23) months. The single NJC case was a 21-years old female patient first diagnosed by an ophthalmologist with corneal deposits, and later received a nephrological evaluation which revealed she had CKD stage 4 (Table 1).
Table 1: Basic clinical and genetic characteristics of the patients diagnosed with NIC or NJC
|
Patient code (sex) |
Clinical diagnosis |
Age of diagnosis |
Years of follow-up |
Height / Age Z score at diagnosis |
Height / Age Z score at last check |
Current condition |
Intra-leukocyte cystine (nm/mg protein) |
Genotype |
|
P1 (M) |
NIC |
11 mo |
1 |
NA |
NA |
Deceased at 20 mo |
5.14 |
ND |
|
P2 (F) |
NIC |
6 mo |
27 |
<-2.0 |
- 4.28 |
Deceased at 28 y.o. |
Higher than cut-offb |
ND |
|
P3 (M) |
NIC |
23 mo |
25 |
NA |
- 2.04 |
Cysteamine-treated |
5.1 |
[g.del57kb] [g.del57kb] |
|
P4 (M) |
NIC |
24 mo |
13 |
-3.32 |
- 3.97GH |
Cysteamine-treated |
2.6 |
[c.992_993insG] [c.992_993insG] |
|
P5 (F)a |
NIC |
20 mo |
8 |
-1.34 |
- 4.2GH |
Cysteamine-treated |
7.14 |
[g.del57kb] [g.del57kb] |
|
P6 (F)a |
NIC |
13 mo |
6 |
0.21 |
- 3.4 |
Cysteamine-treated |
ND |
[g.del57kb] [g.del57kb] |
|
P7 (F) |
NIC |
13 mo |
5 |
-2.46 |
-1.54 |
Cysteamine-treated |
ND |
[g.del57kb] [g.del57kb] |
|
P8 (F) |
NJC |
21 y.o. |
0.8 |
-3.03 |
- 3.03 |
Cysteamine-treated |
ND |
[c.416C>T] [c.416C>T] |
|
P9 (F) |
NIC |
28 mo |
0.1 |
-4.29 |
-4.29 |
Cysteamine recently started |
ND |
[g.del57kb] [g.del57kb] |
NIC, nephropathic infantile cystinosis. NJC, nephropathic juvenile cystinosis. NA, not available, ND, not determined a These patients are siblings. bOriginal report with the exact value was not available, but medical records indicated it was higher than the cut-off. GH Growth Hormone
The two NIC patients diagnosed before 2000 died during the follow-up. The first one (P1) died when he was 20 months old because of hypovolemic shock due to gastrointestinal losses coupled with polyuria. The second one (P2) died when she was 28 years old after respiratory failure due to restrictive lung disease apparently related to a cystinosis-induced myopathy given the long course of the disease.
Time to diagnosis and cysteamine therapy
Diagnosis of cystinosis was established by intra-leukocyte cystine values above the cut-off point defined by most laboratories (>1 nmol half-cystine/mg of protein) in 5 patients (Table 1). The diagnosis in the remaining cases was confirmed by CTNS genetic analysis. The median time elapsed between cystinosis suspicion to confirmation of diagnosis was 6.5 (range 1 to 10) months, while the posterior median time elapsed between the diagnosis confirmation and the cysteamine initiation was 2 (range 1 to 79) months (Table 2).
Table 2: eGFR and cysteamine therapy of the patients diagnosed with NIC or NJC
|
Patient code (sex) |
Interval between diagnosis and cysteamine initiation (months)
|
Cysteamine therapyb |
eGFR at diagnosis (ml/min/1.73) |
eGFR at last check (ml/min/1.73) |
Kidney Transplant |
|
P1 (M) |
5 |
According to consensus |
0.5 mg/dL (creatinine)c |
0.73 mg/dL (creatinine)c |
No |
|
P2 (F) |
79 |
Late access and half of the recommended dose |
20 |
25.6 |
Living donor at 6 y.o |
|
P3 (M) |
2 |
Half of the recommended dose until KT |
44 |
139 |
Cadaveric donor at 9 y.o |
|
P4 (M) |
2 |
According to consensus |
64 |
37.4 |
No |
|
P5 (F)a |
2 |
According to consensus |
97 |
24 |
No |
|
P6 (F)a |
3 |
According to consensus |
87 |
51 |
No |
|
P7 (F) |
1 |
According to consensus |
27 |
35 |
No |
|
P8 (F) |
1 |
Late and irregular access |
23 |
14.4 |
No |
|
P9 (F) |
2 |
According to consensus |
56 |
56 |
No |
a These patients are siblings. b Consensus describes cysteamine therapy as 1.30 g/m2/day for children <12 years and 2 g/day for patients >12 years if weight is >50 kg, divided 4 times each day or 60 a 90 mg/k/day 4 times each day. c Height data to estimate eGFR was missing.
Kidney outcomes and extrarenal manifestations
We analyzed the eGFR of 7 out of the 8 NIC patients throughout their follow-ups and compared their first and last values (Figure 1, Table 2). Patient P1 is not shown because clinical information was missing.
Figure 1:
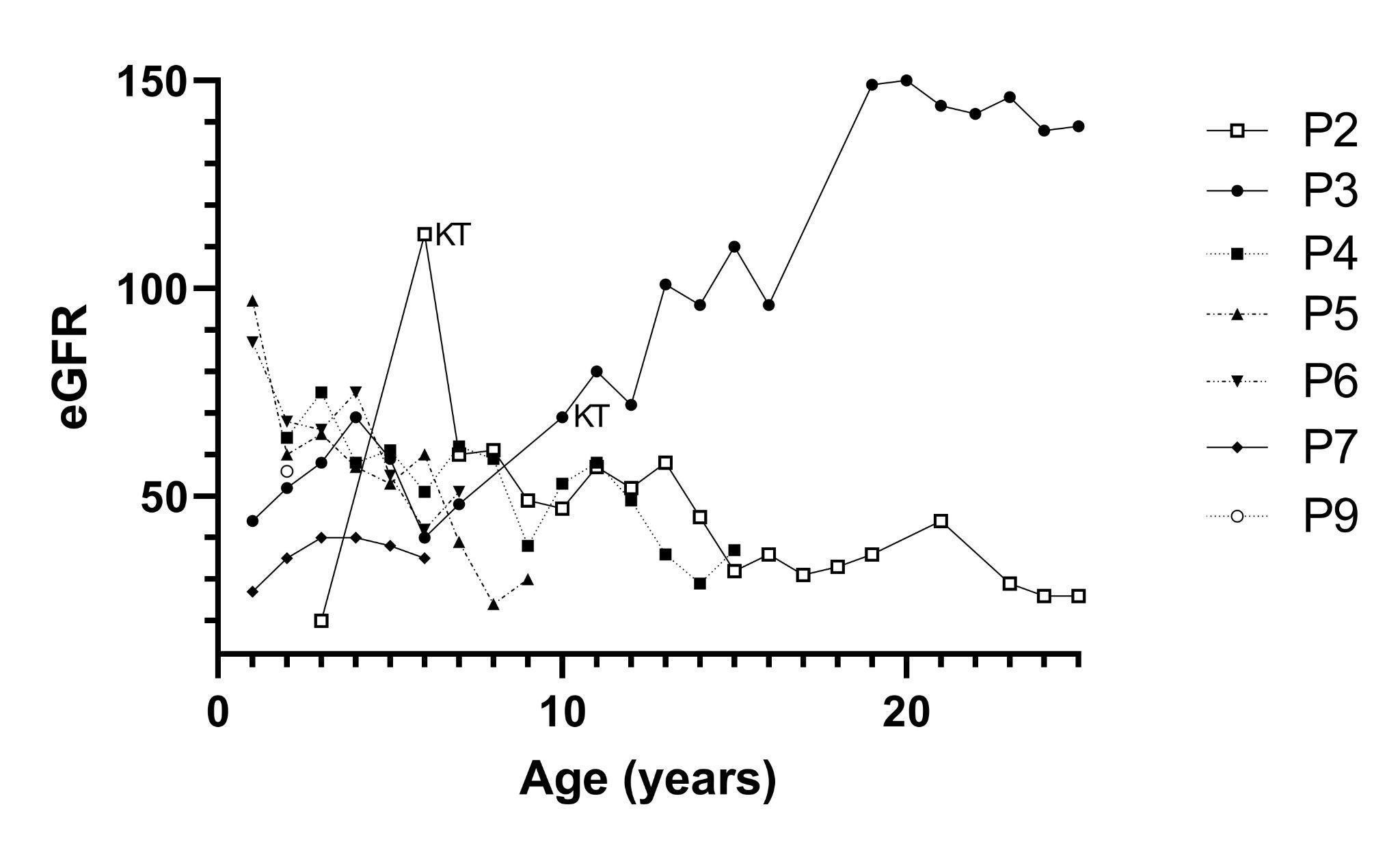
Data of patient P2 was obtained at the age of 3 when she started on dialysis, receiving a kidney allograft 3 years later. Later on, she had a decline in allograft function. During her life, she had late and irregular access to cysteamine and developed multi-organic complications related to cystinosis. Patient P3 is an adult male patient who underwent KT at 9 years of age. He continued cysteamine therapy and has a normal creatinine clearance currently. P4 was diagnosed at the age of two and two months later he started cysteamine therapy according to consensus. His eGFR at the first visit was 64 ml/min/1.73m2 and 13 years later, at the end of the follow-up period, his eGFR declined to 37.4 ml/min/1.73m2. Two siblings (P5 and P6) showed normal kidney function at the beginning with a progressive decline in the follow-up. Patient P7 showed an eGFR of 27 ml/min/1.73m2 at diagnosis that improved slightly after starting cysteamine therapy. P8 was the single NJC case, a 21-year-old female patient, who received irregular cysteamine therapy and rapidly reached CKD stage 5. Patient P9 was diagnosed recently, therefore, only her initial evaluation is shown. At the last visit, the remaining five NIC living patients were classified in CKD stages 3-4.
When tubulopathy and CKD treatments were provided, all patients improved their clinical and biochemical values. Indomethacin was indicated in the siblings to decrease polyuria, but it was stopped after 2 years because eGFR declined. The remaining patients were able to maintain hydric balance with oral intake.
Regarding extrarenal manifestations, all patients developed ocular involvement with corneal deposits of cystine crystals and started pharmacy-prepared cysteamine eye drops permanently. Seven patients had mild photophobia which improved in one of them after KT. Five patients developed hypothyroidism requiring levothyroxine treatment. Three patients (P2, P3 and P4) that had been monitored for more than 10 years developed other extrarenal complications such as central nervous system involvement, myopathies, swallowing difficulties, hypogonadism and diabetes mellitus.
The single NJC patient received low and erratic cysteamine doses obtained by donations and reached CKD stage 5 less than one year after diagnosis.
Growth
We analyzed the height-for-age Z-scores throughout the follow-ups of NIC patients (Figure 2). At the time of diagnosis, only one case had a normal Z-score and five cases had Z-scores below -2.0. Three patients (P3, P4 and P7) improved their height during the first three years, but two of them (P3 and P4) deteriorated later over time. In contrast, siblings P5 and P6 showed initial Z-scores close to normal limits and although they had access to cysteamine their Z-scores worsened in the following years. The use of rhGH was prescribed to three patients, however, one of them (P6) has not been able to start due to financial issues.
Figure 2
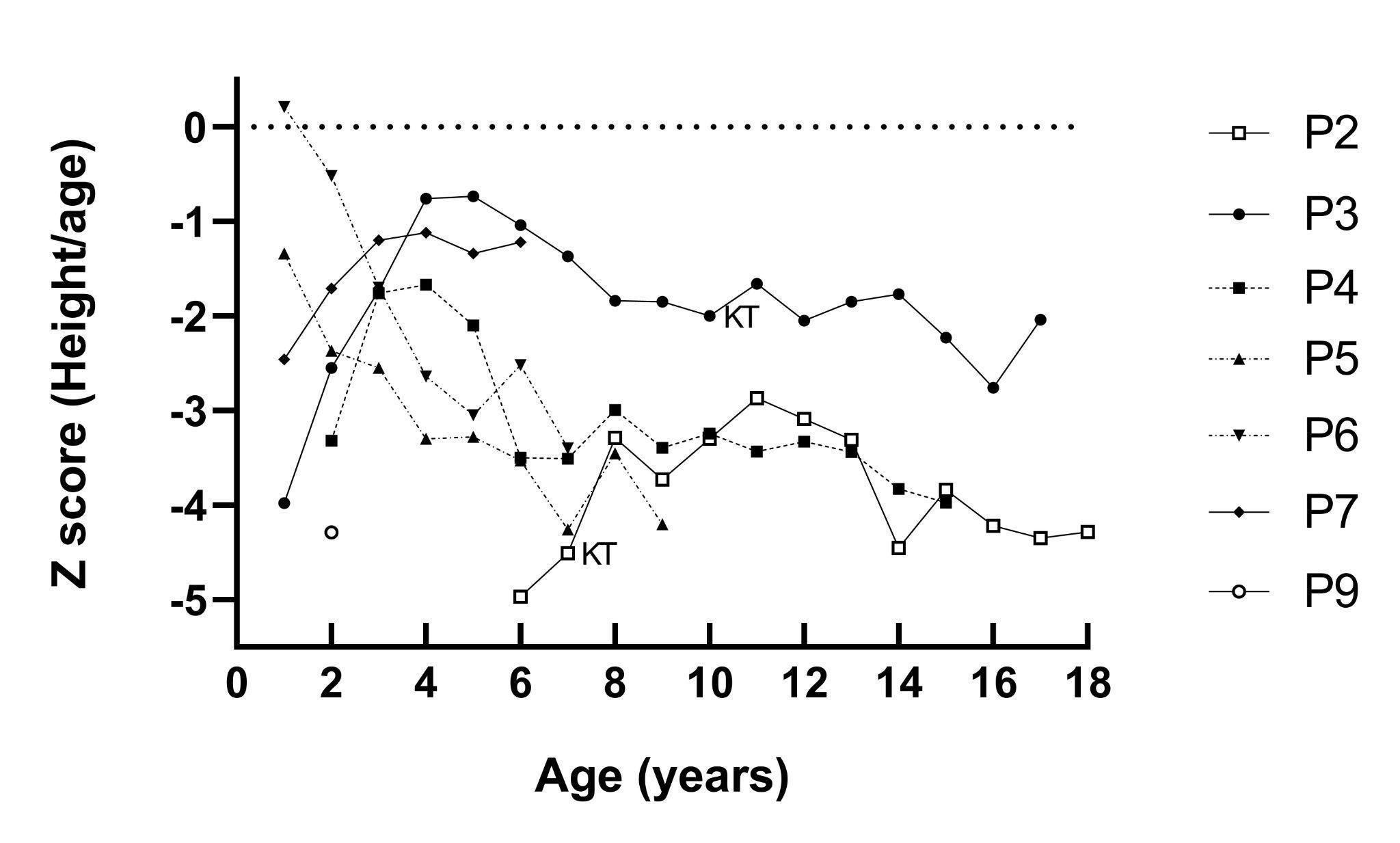
At diagnosis, three patients (P1, P3 and P5) were severely underweight. P1 died before nutritional intervention could be performed. None of the patients underwent gastrostomy. During the follow-up, all patients reached normal weight for height except P4 who was at underweight risk at the age of 9 and got worse at 13 years old with body mass index for-age Z-score of -1.31 and -3.28, respectively. In addition, hypogonadism was diagnosed during this period.
Genetic Findings
Genetic analysis was performed on 7 out of the 9 patients, given that 2 patients died before implementing molecular diagnostic methods. The results confirmed a NIC or NJC diagnosis in all cases. In the first approach to test the presence of the del57kb variant, 5/7 (71%) cases were homozygous del57kb carriers (Table 1, Figure 3).
Figure 3
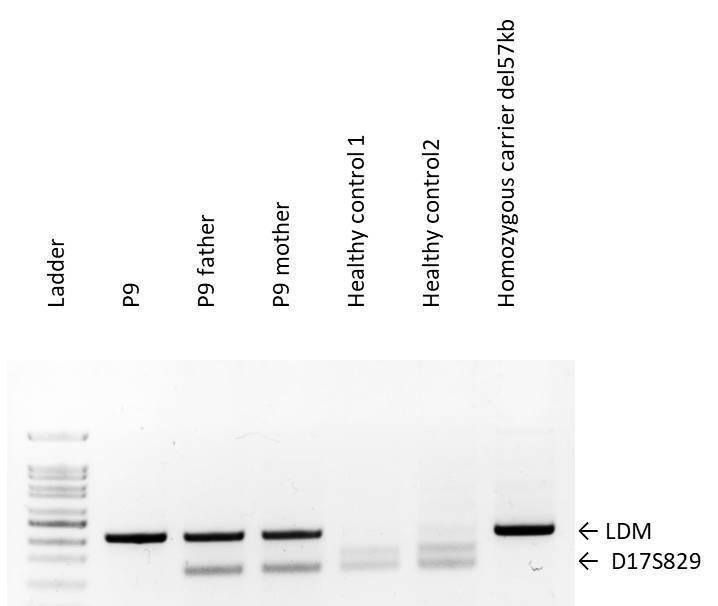
The two remaining patients required sequencing of the CTNS coding exons. We identified in patient P4 a homozygous nucleotide insertion in exon 12 (c.992_993insG) that predicts a reading frame disruption in the last amino acids (p.Asp332ArgfsX33), yielding a protein slightly shorter than the normal. Patient P8, diagnosed with NJC in adulthood, carried a homozygous nucleotide substitution in exon 7 (c.416C>T) that predicted a likely pathogenic amino acid substitution (p.Ser139Phe) (Figure 4).
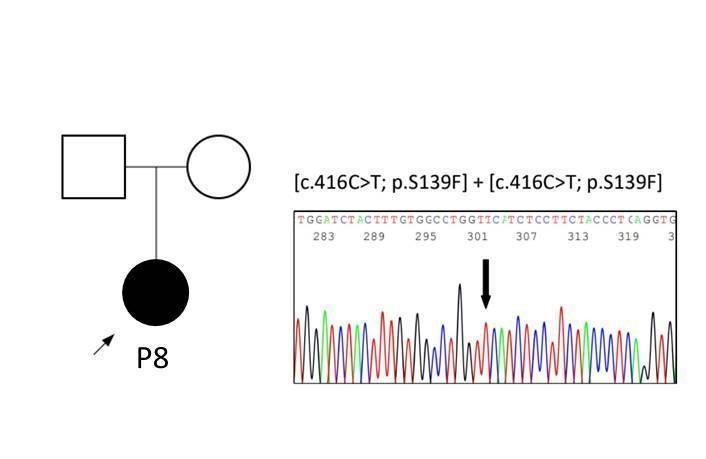
Figure 4
This variant has been described previously in NJC cases and functional assays showed abolished cystine transport.
Discussion
To the best of our knowledge, this is the first report to describe the clinical and genetic characteristics of nephropathic cystinosis patients in a Latin American country along with their long-term follow-up. We collected data for 9 patients in the last 27 years, which was less than expected based on the birth rate in Chile during this period. Our data shows a prevalence of 0.36 per million people, while the worldwide prevalence is 1.6 per million people. This difference could be explained by fewer defective CTNS alleles in Chile, although misdiagnosis or underdiagnosis cannot be ruled out since the predominant infantile presentation has a silent period during gestation and the first semester of life4,20,21. Additionally, physicians often lack the experience to perform diagnoses and provide adequate clinical management.
The median age at NIC diagnosis was 16.5 months, similar to the results in other developing or developed countries11. The single NJC case was a woman diagnosed by an ophthalmologist, and a subsequent evaluation showed she had CKD stage 4, suggesting cystinosis might have started during adolescence without evident symptoms. Our patients received a late diagnosis, which is partly explained by the late recognition of symptoms. Early detection is critical and if treatment is started immediately after birth, progression might be reduced or even prevented21,22. The most cost-efficient approach to prevent multi-organ damage might be a newborn next-generation sequencing (NGS) screening program to identify CTNS variants23,24. Limited resources in some countries restrict access to NGS even though it might improve outcomes in medically actionable genetic conditions25.
Previous reviews describe a clinical course of cystinosis similar to the cases reported here8,26. Two of the patients diagnosed before 2000 had difficulties establishing their diagnosis and receiving cysteamine. Both required KT in the first decade after diagnosis with optimal graft survival in one of them. In contrast, the last two NIC-diagnosed patients had an early genetic diagnosis and presented stable eGFR in their short-term follow-up. Moreover, P7 showed a slight improvement from CKD stage 4 to 3 after two years of therapy. The drop in eGFR in the remaining patients within the first years drew our attention; even P5, who had prompt access to cysteamine since she was the younger sister of P6, who was diagnosed one year before. This could be a consequence of non-adherence since drug tolerance was suboptimal for the siblings whose mother reported a high frequency of vomiting. Gastrostomy was recommended, but the family rejected the procedure.
All patients had ophthalmologic involvement and cysteamine eye drops were indicated, however, a substantial improvement was not observed. Commercially ophthalmic solutions containing cysteamine hydrochloride (0.55%) eye drops are not available in Chile. Therefore, parents request their preparation with prescription formulas in pharmacies without any regulation to assure their quality and effectiveness.
Growth impairment is frequently observed in cystinosis because of tubulopathy losses and CKD. Our patients' Z-scores were severely impacted as CKD progressed, including those cases treated with rhGH. For instance, P4 presented hypogonadism that could have interfered with growth and muscle mass gain, among other metabolic alterations. In Chile, rhGH is provided either by the public or private health systems, but it is guaranteed only when pediatric patients have reached CKD stage 4.
NIC is considered a progressive disease and cystine-depleting therapies have extended patients' lifespan beyond 40 years of age. Previous studies in other cohorts describe a mortality that ranges between 10% and 33%, but an adequate administration of cysteamine is associated with lower mortality rate27. In our cohort 2 out of 9 patients died, which is higher than the mortality described in Latin American countries28. Digestive losses represent a high risk of morbimortality, and it was the cause of death in P1. Patient P2 underwent peritoneal dialysis and KT during childhood but she had irregular cysteamine therapy during her life, which might have contributed to the development of multi-organic complications and death.
Two additional patients also received late and/or irregular treatment in the early stages. Most patients received doses according to the Spanish consensus, but disparities in how to get cysteamine were observed26.
Cysteamine is neither commercially available nor financed in Chile. Like other orphan drugs, it is imported as an individualized request, increasing costs. Furthermore, cysteamine has a financial cost of approximately 1.5 times the minimum wage, resulting in restrictive access for most patients. Some difficulties in therapy access come from the Chilean health system. This is a dual model with the public (FONASA, National Health Fund) and private (ISAPREs) components that provide insurance and health services with nearly 80% of the population currently enrolled in FONASA29. There are no clear definitions or specific laws in Chile for rare diseases such as cystinosis, impeding the therapies coverage30. Access to therapy is achieved after legal actions against the state or private system in courts of justice. Most patients in our cohort belong to FONASA, but even the two patients from ISAPREs had difficulties obtaining the drug at appropriate dosages. Over the last 23 years, most patients achieve regular cysteamine funding up to 12 months after initiating legal actions. In the meantime, some families had to pay for treatment by themselves or receive donations from other families which partially supply the drug requirement and this period without appropriate therapy increases the multiorgan involvement27.
Extrarenal manifestations such as hypothyroidism, hypogonadism, central nervous system involvement, myopathies, swallowing difficulties and diabetes mellitus have been described in the literature26, due to the accumulation of cystine in different organs. In our cohort, 5 patients developed hypothyroidism. Among them, 3 patients also developed several other extrarrenal complications. P2 had the most severe complications besides CKD, including diabetes mellitus, swallowing difficulties and myopathies, which contributed to her death at 28 years old. She was diagnosed with cystinosis at a time when it was extremely difficult to obtain cysteamine in our country, and was started on this therapy approximately 7 years after the diagnosis. Furthermore, due to financial difficulties, she received insufficient doses of cysteamine.
Adherence to cysteamine therapy is a major problem associated with less favorable outcomes. Intra-leukocyte cystine quantification to monitor adherence or dose adequacy is not available in Chile and requires fresh blood samples that must arrive in less than 24 hours to overseas laboratories. Therefore, our patients receive the weight-based or body surface area recommended cysteamine doses.
The genetic analysis yielded a 100% mutation detection rate, and 5/7 cases were homozygous del57kb carriers. LDM/D17S829 PCR emerges as a cost-effective diagnostic method in comparison to measuring intra-leukocyte cystine. Case P7 was the first Chilean case genetically tested in 2016 and harbored the del57kb variant, frequently detected in Europe31. Currently, Chilean population originated from the admixture between Native Amerindians and Europeans migrating since the XV century32. Rare recessive diseases are frequently associated with endogamy, and all our patients were homozygous carriers. Although most consanguineous marriages in Chile occur among first cousins, none of the participants declared consanguinity before second-degree relatives33. We confirmed a heterozygous carrier status in all the parents that requested testing, which facilitates genetic counseling for forthcoming gestations. This information may also be considered for living donor studies in the future.
The novel allele in exon 12 might be a variant of European origin or might have arisen in Native Amerindians or Asians before settlement in America. The single NJC patient carried a variant in exon 7 of Spanish origin, compatible with the known European ancestry among Chileans34. Analyses of CTNS variants in other Latin American countries, particularly Mexico and Brazil, have shown that one-third of all cases carry the del57kb variant35,36. Although our dataset is limited and recruitment might be biased, we propose to assay first the presence of del57kb and, if negative or only one variant is detected, sequence all CTNS exons in cystinosis-suspicious patients.
The impact of different variants as determinants of clinical evolution has not shown a difference between types of CTNS mutations and kidney survival8,36,37. In our cohort, the truncating del57kb variant was exclusively found in infantile cystinosis, suggesting it might be associated with the early development of symptoms. However, carriers of CTNS truncating mutations with late CKD stage 5 have been reported worldwide, highlighting that opportune diagnosis and precocious cysteamine therapy are key factors to improve kidney survival, while other therapies and potential cures continue being developed36-38.
Conclusions
Developing countries face many disparities in providing NIC/NJC patients with adequate healthcare. Cystinosis is a disorder still poorly known by the medical community in Chile, without official statistics and treatment support from the health system. In this study, we set up a collaborative research group to spread clinical and diagnostic aspects to the medical and patient/parent community. We think that these findings will improve multidisciplinary management and kidney survival.
Sharing this evidence with health authorities might contribute to guaranteeing treatment access and accelerating the diagnostic approach through del57kb detection. The clinical-exome sequencing strategy for variant identification has shown to be clinically useful in rare diseases such as cystinosis and is expected to be gradually implemented worldwide, if the costs continue declining while researchers and physicians improve their training. Opportune genetic testing may facilitate early diagnosis and cysteamine treatment, known to be associated with better outcomes. Financed evidence-based standardized protocols are required to generate public policies to benefit patients with rare diseases such as nephropathic cystinosis.
Declarations
Funding: Not applicable.
Conflict of interest: The authors declare no conflict of interest.
Availability of data and material: Not applicable.
Code availability: Not applicable.
A preprint was posted (June 2022) on https://www.researchsquare.com/article/rs-1717027/v1
Authors' contributions: P.K., J.G., L.B. and J.L.G. have conceived and designed the study contributing equally to this work. All authors provided intellectual content, revised the drafts and approved the final version.
References
- Gahl WA, Bashan N, Tietze F, et al. (1982). Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science 217: 1263-1265. https://doi: 10.1126/science.7112129.
- Attard M, Jean G, Forestier L, et al. (1999). Severity of phenotype in cystinosis varies with mutations in the CTNS gene: predicted effect on the model of cystinosin. Hum Mol Genet 8: 2507-2514. https://doi: 10.1093/hmg/8.13.2507
- Broyer M, Guillot M, Gubler MC, et al. (1981). Infantile cystinosis: a reappraisal of early and late symptoms. Adv Nephrol Necker Hosp 10: 137-166.
- Gahl WA, Thoene JG, Schneider JA et al. (2002). Cystinosis. N Engl J Med 347: 111-121. https://doi: 10.1056/NEJMra020552.
- Markello TC, Bernardini IM, Gahl WA, et al. (1993). Improved renal function in children with cystinosis treated with cysteamine. N Engl J Med 328: 1157-1162. https://doi: 10.1056/NEJM199304223281604.
- Wilmer MJ, Schoeber JP, van den Heuvel LP, et al. (2011). Cystinosis: practical tools for diagnosis and treatment. Pediatr Nephrol 26: 205-215. https://doi: 10.1007/s00467-010-1627-6.
- Levtchenko E, van den Heuvel L, Emma F, et al. (2014). Clinical utility gene card for: cystinosis. Eur J Hum Genet 22. https://doi: 10.1038/ejhg.2013.204.
- Topaloglu R (2021). Nephropathic cystinosis: an update on genetic conditioning Pediatr Nephrol 36:1347-1352. https://doi: 10.1007/s00467-020-04638-9.
- Nesterova G, Gahl W (2008). Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr Nephrol 23:863-878. https://doi: 10.1007/s00467-007-0650-8.
- Bertholet-Thomas A, Bacchetta J, Tasic V, et al. (2014). Nephropathic cystinosis--a gap between developing and developed nations. N Engl J Med 370:1366-1367. https://doi: 10.1056/NEJMc1309480.
- Bertholet-Thomas A, Berthiller J, Tasic V, Kassai B, et al. (2017). Worldwide view of nephropathic cystinosis: results from a survey from 30 countries. BMC Nephrol 18:210. https://doi: 10.1186/s12882-017-0633-3.
- Martinez-Millana A, Hulst JM, Boon M, Witters P, et al. (2018). Optimisation of children z-score calculation based on new statistical techniques. PLoS One 13(12): e0208362. https://doi: 10.1371/journal.pone.0208362.
- Schwartz GJ, Muñoz A, Schneider MF, et al. (2009). New equations to estimate GFR in children with CKD. J Am Soc Nephrol 20: 629-637. https://doi: 10.1681/ASN.2008030287.
- Levey AS, Stevens LA, Schmid CH, et al. (2009). A new equation to estimate glomerular filtration rate. Ann Intern Med 150:604-612. https://doi: 10.7326/0003-4819-150-9-200905050-00006.
- Anikster Y, Lucero C, Touchman JW, et al. (1999). Identification and detection of the common 65-kb deletion breakpoint in the nephropathic cystinosis gene (CTNS). Mol Genet Metab 66(2): 111-6. https://doi: 10.1006/mgme.1998.2790.
- Kent WJ, Sugnet CW, Furey TS, et al. (2002). The human genome browser at UCSC. Genome Res 12(6): 996-1006. https://doi: 10.1101/gr.229102.
- Schwarz JM, Cooper DN, Schuelke M, et al. (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11(4): 361-2. https://doi: 10.1038/nmeth.2890.
- Sim NL, Kumar P, Hu J, Henikoff S, et al. (2012). SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 40 (Web Server issue): W452-7. https://doi: 10.1093/nar/gks539.
- Adzhubei IA, Schmidt S, Peshkin L, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods7(4): 248-9. https://doi: 10.1038/nmeth0410-248.
- Broyer M, Niaudet P (2012). Cystinosis. In: Saudubray, JM., van den Berghe, G., Walter, J.H. (eds) Inborn Metabolic Diseases. Springer, Berlin, Heidelberg. pp617-624. https://doi: 10.1007/978-3-642-15720-2_43.
- Elmonem MA, Veys KR, Soliman NA, et al. (2016). Cystinosis: a review. Orphanet J Rare Dis 11: 47. https://doi: 10.1186/s13023-016-0426-y.
- Bäumner S, Weber LT (2018). Nephropathic Cystinosis: Symptoms, Treatment, and Perspectives of a Systemic Disease. Front Pediatr 6:58. https://doi: 10.3389/fped.2018.00058.
- Hohenfellner K, Bergmann C, Fleige T, et al. (2019). Molecular based newborn screening in Germany: Follow-up for cystinosis. Mol Genet Metab Rep 21: 100514. https://doi: 10.1016/j.ymgmr.2019.100514.
- Fleige T, Burggraf S, Czibere L, et al. (2020). Next generation sequencing as second-tier test in high-throughput newborn screening for nephropathic cystinosis. Eur J Hum Genet 28: 193-201. https://doi: 10.1038/s41431-019-0521-3.
- Parad RB, Kaler SG, Mauceli E, et al. (2020). Targeted next generation sequencing for newborn screening of Menkes disease. Mol Genet Metab Rep 24: 100625. https://doi: 10.1016/j.ymgmr.2020.100625.
- Ariceta G, Camacho JA, Fernández-Obispo M, et al. (2015). Cystinosis in adult and adolescent patients: Recommendations for the comprehensive care of cystinosis. Nefrologia 35: 304-321. https://doi: 10.1016/j.nefroe.2015.06.010.
- Gahl WA, Balog JZ, Kleta R. (2007). Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann. Intern. Med. 147(4): 242-50. https://doi:10.7326/0003-4819-147-4-200708210-00006.
- Vaisbich MH, Koch VH (2010). Report of a Brazilian multicenter study on nephropathic cystinosis. Nephron Clin Pract 114: c12-18. https://doi: 10.1159/000245065.
- Oliveira SC, Machado CV, Hein ARA, et al. (2021). Public-private relations in Chile's health system: regulation, funding and service delivery. Cien Saude Colet 26:4529-4540. https://doi: 10.1590/1413-812320212610.09892021.
- Encina G, Castillo-Laborde C, Lecaros JA, et al. (2019). Rare diseases in Chile: challenges and recommendations in universal health coverage context. Orphanet J Rare Dis 14: 289. https://doi: 10.1186/s13023-019-1261-8.
- Krall P, Nualart D, Grandy J (2018). Nephropathic cystinosis: Report of one case. Rev Med Chil 146: 111-115. https://doi: 10.4067/s0034-98872018000100111.
- Eyheramendy S, Martinez FI, Manevy F, et al. (2015). Genetic structure characterization of Chileans reflects historical immigration patterns. Nat Commun 6: 6472. https://doi: 10.1038/ncomms7472.
- Blanco R, Chakraborty R (1975). Consanguinity and demography in some Chilean populations. Hum Hered 25: 477-487. https://doi:10.1159/000152763.
- Macías-Vidal J, Rodés M, Hernández-Pérez JM, et al. (2009). Analysis of the CTNS gene in 32 cystinosis patients from Spain. Clin genet 76: 486-489. https://doi: 10.1111/J.1399-0004.2009. 01222.X.
- Alcántara-Ortigoza MÁ, Belmont-Martínez L, Vela-Amieva M, et al. (2008). Analysis of the CTNS gene in nephropathic cystinosis Mexican patients: Report of four novel mutations and identification of a false positive 57-kb deletion genotype with LDM-2/exon 4 multiplex PCR assay. Genet Test 12: 409-414. https://doi: 10.1089/gte.2008.0014.
- Topaloglu R, Gulhan B, Ä°nözü M, et al. (2017). The Clinical and Mutational Spectrum of Turkish Patients with Cystinosis. Clin J Am Soc Nephrol 12: 1634–1641. https://doi: 10.2215/CJN.00180117.
- Emma F, Hoff WV, Hohenfellner K, et al. (2021). An international cohort study spanning five decades assessed outcomes of nephropathic cystinosis. Kidney Int 100: 1112-1123. https://doi: 10.1016/j.kint.2021.06.019.
- Cherqui S (2021). Hematopoietic Stem Cell Gene Therapy for Cystinosis: From Bench-to-Bedside. Cells 10: 3273. https://doi: 10.3390/cells10123273.