
Congenital Generalized Lipodystrophy
Josivan Gomes Lima1*, Marcel Catão Ferreira dos Santos1, Julliane Tamara Araújo de Melo Campos2
1Departamento de medicina clínica, disciplina de endocrinologia e metabologia. Hospital Universitário Onofre Lopes, Universidade Federal do Rio Grande do Norte (UFRN), Natal, RN, Brazil
2Faculty of Health Sciences of Trairi, Federal University of Rio Grande do North (UFRN), Natal, RN, Brazil
Abstract
Congenital Generalized Lipodystrophy (CGL) is a rare and severe autosomal recessive disease. Patients are defective in the storage of body fat and, consequently, they deposit fat in ectopic tissues, mainly liver, and can develop cirrhosis. Insulin resistance is a typical finding, causing diabetes that require high daily doses of insulin. In the state of Rio Grande do Norte, Brazil, we have one of the largest cohorts of patients with CGL. In this article, we review pathophysiology, clinical picture and treatment of this disease.
Introduction
Type 2 diabetes is a world health problem, and usually results from excessive weight and increased visceral fat causing peripheral insulin resistance and an inability of the pancreas to release insulin to compensate this resistance. Other less common types of diabetes occur due to specific genetic mutations, like the Congenital Generalized Lipodystrophy (CGL), also known as Berardinelli-Seip Congenital Lipodystrophy (BSCL). CGL is an autosomal recessive disease that is classified into four types, based on gene mutation. The altered genes play essential functions for adipocyte formation, lipid production and proper storage inside the adipocyte. The mutations decrease adipose tissue with consequent deposition of fat in ectopic sites, causing fat liver, altered carbohydrate metabolism, severe insulin resistance with hyperinsulinemia and acromegaloid features, and dyslipidemia1-3. The CGL syndrome has around 500 cases reported in the world. In Brazil, in the State of Rio Grande do Norte (RN), we have diagnosed, treated, and followed 54 cases in the past 20 years4, 5. In a descriptive study using secondary data, we estimated a total of 103 patients in RN6. This indicates a much higher prevalence than that reported in the literature (1: 1 million)7.
Triacylglycerol formation and storage in lipid droplets
The biosynthesis of triglycerides and phospholipids (Figure 1A) starts with glycerol-3-phosphate acyltransferase (GPAT) acylating the glycerol-3-phosphate in position 1, forming 1-Acylglycerol-3-phosphate (lysophosphatidic acid). It is followed by another acylation step at position two by the enzyme AGPAT (1-Acylglycerol-3-phosphate acyltransferase), originating 1,2-Diacylglycerol-3-phosphate (phosphatidic acid). It is a key intermediate step in the biosynthesis pathway of both triglycerides and phosphoglycerides. There are 11 isoforms of AGPAT enzymes, encoded by different genes4. AGPAT1 and AGPAT2 are the most extensively studied. AGPAT1 is present at high levels in testis, pancreas, and, to a lesser extent, in adipose tissue and other tissues like heart, placenta, brain, lung, whereas AGPAT2 is abundant in fatty tissue. In the following steps, the cytosolic enzyme phosphatidic acid phosphatase (PAP or lipin) originates 1,2-diacylglycerol, and the 1,2-diacylglycerol acyltransferase (DGAT) forms triacylglycerol4. Phosphatidic acid and diacylglycerol can also originate other phospholipids such as cardiolipin, phosphatidylinositol, and phosphatidylcholine.
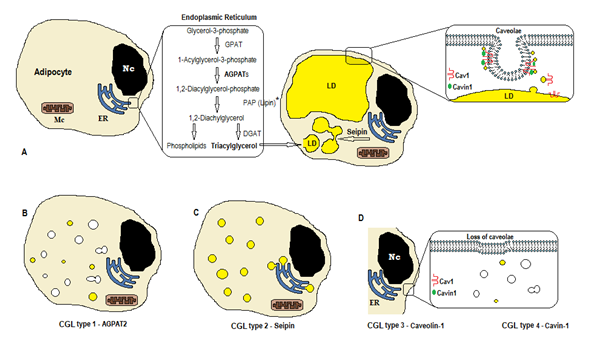
Figure 1. Scheme of triglycerides synthesis according to CGL types. (A) Normal synthesis and storage of triacylglycerol (TAG) in the adipocyte. (B) Mutation of AGPAT2 decreases TAG production (some is still synthesized under stimulation of other AGPATs). (C) Mutation of seipin gene decrease TAG synthesis and lipid droplet (LD) formation and fusion. (D) Caveolin-1 and Cavin-1 are required for the formation and stabilization of the caveolae. Mutation in CAV1 (type 3) or CAVIN1 (type 4) can cause loss of caveolae in the membrane. Nc, nucleus. ER, endoplasmic reticulum. Mc, mitochondria. *Lipin is a cytosolic enzyme anchored by seipin in the ER.
Those reactions occur in the adipocytes’ endoplasmic reticulum (ER), where a progressive accumulation of triglycerides causes the formation of small lipid droplets (LD)8. The product of the gene BSCL2 is a transmembrane protein called seipin that causes the fusion of small LD, originating large LD. Seipin resides in the ER and concentrates at the junction with nascent LD, facilitating the lipid traffic between ER and LD and the incorporation of triglycerides in LD9. Seipin may also act as an ER anchor to the cytosolic enzyme lipin 1. Besides being necessary for lipid droplet fusion, size, and morphology, seipin is also essential for adipogenesis (via interaction to lipin 1) and cellular triglyceride lipolysis10, 11. Deficiency of seipin does hamper the differentiation of pre-adipocytes to adipocytes and affects the final maturation9, as shown by studies in mesenchymal stem cells with BSCL2 knocked out12. Non-adipose tissues also express seipin, and other functions are to be determined.
In the adipocytes, caveolae, which are specialized 50-100nm membrane invaginations, account for 20% of the plasma membrane area, making the adipocytes the cells with the highest density of caveolae13. The formation of lipid droplets needs a membrane protein (Caveolin - the main component of caveolae membranes) and a cytoplasmic protein (Cavin-1)14. The genes CAV1, CAV2, and CAV3 encode three forms of caveolin with similar structures (Caveolin-1, Caveolin-2, and Caveolin-3, respectively). Caveolin-1 and Caveolin-2 are present in adipocytes, fibroblast, and endothelial cells, and Caveolin-3 is present only in skeletal and cardiac muscle13, 15. Caveolin-1 is the most important and the most studied. It is expressed in two different isoforms (1a and 1b). Caveolin-1 translocates from the plasma membrane to lipid droplet, being necessary to lipid trafficking and metabolism16. Lipid droplets store triglycerides after feeding and these molecules are hydrolyzed to fatty acid, and released during fasting; this mechanism may be regulated by Caveolin-116. Caveolin-1 deficiency also increases susceptibility to cell death by autophagy17.
The gene CAVIN1 encodes a cytoplasmic protein called caveolae associated protein 1 (Cavin-1)14, 16, that is obligatory for the formation and stabilization of caveolae. Cavin-1 is expressed in adipocytes, muscle cells, and other cells, and is also essential in the transmission of caveolae-originated signals14, 18. Knockout of the CAV1 gene causes a lack of caveolae in non-muscle cells, whereas the knockout of CAVIN1 causes the absence of caveolae in all tissues, including muscle14. The lack of caveolae can affect regulation of lipolysis, fatty acid flux, triglyceride synthesis, and the signals of other pathways.
Types of CGL
Based on detectable genetic alterations, four types are described. Types 1 and 2 are responsible for over 95% of cases, and type 2 has a more severely affected phenotype. Only one case of type 3 and around 30 cases of type 4 have been reported4.
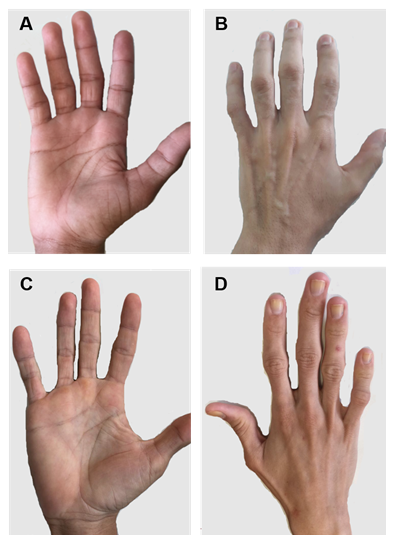
Figure 2. Hands of patients with CGL types 1 and 2. (A) and (B) Anterior and posterior views of hands of type 1 patients. Apparently normal hands, since there is still mechanical fat tissue. (C) and (D) Anterior and posterior views of hands of type 2 patients. The severity of the disease is greater, and the lack of fat is evident and easily noticeable.
CGL Type 1. In 1999, Garg et al. described patients’ mutation on chromosome 9q34, and three years later Agarwal et al. showed AGPAT2 as the enzyme affected by this mutation2, 19. Due to mutation of this AGPAT2, none or minimal production of triacylglycerol happens by the stimulus of other isoforms. The phenotype of AGPAT2 knockout mice is similar to that of humans with CGL type, confirming the role of this enzyme in the pathophysiology20, 21.
CGL Type 2. Magre et al. were the first to identify the mutation in the seipin gene (chromosome 11q13)3. Mutations (mostly nonsense) of the seipin gene (BSCL2) produce a truncated protein and can affect lipid metabolism by different mechanisms: a) decrease in seipin stability; b) reduction in ability to bind lipin 1; and c) failure to oligomerize and localize itself exclusively to the ER membrane11. Some cells are still able to generate triacylglycerol and small lipid droplets, but large lipid droplets are absent due to loss of the ability of fusion of these small lipid droplets. There is also a failure in the expression of adipogenic factors, such as the peroxisome proliferator-activated receptor gamma (PPARG), as well as adiponectin and adipocyte fatty acid binding protein (FABP4)11, 16. Seipin deficiency impairs adipogenesis, increases lipolysis, and prevents triglycerides accumulation in adipocytes.
CGL type 3. This type was described recently in a patient who despite having CGL phenotype did not have mutations in genes AGPAT2 or BSCL222. Mice with a mutation in Cav1 are resistant to diet-induced obesity and have insulin resistance, hypertriglyceridemia, decreased adiponectin, reduced fat mass, and small adipocytes16. After choosing candidate genes based on studies in mice, Kim et al. confirmed the presence of a nonsense mutation in the caveolin-1 gene (CAV1), on chromosome 7q3122.
CGL type 4. In this is a rare type the affected gene is the CAVIN1, which encodes the protein Cavin-1. In humans, it has been reported in patients with generalized congenital lipodystrophy and muscular dystrophy15, 23.
Recently, mutations in the PCYT1A and PPARG genes have also been described causing lipodystrophy24, 25.
Clinical features
CGL patients usually present acromegaloid facies, acanthosis nigricans, phebomegaly, hepatomegaly, and muscular hypertrophy5, 26, 27. Several authors cite umbilical hernia as a clinical finding of the syndrome26. We evaluated the frequency of it in our series of patients, and none of them presented this change28. In fact, the absence of periumbilical adipose tissue causes protrusion of the umbilical scar, and this may be mistakenly diagnosed as a hernia28, 29.
Once adipocytes cannot adequately store fat, it accumulates in other tissues such as liver, and muscles, causing severe insulin resistance. Bone densitometry (DXA) may show normal or high bone mineral density30 and reduced total body fat (usually lower than 6%)27. As a consequence of low body fat, serum adiponectin and leptin are low too27. As leptin is essential in controlling hunger, these patients typically have hyperphagia, which is readily apparent since childhood. Adiponectin plays an important role as an insulin sensitizer, and its lack worsens the insulin resistance. Despite this, initially, glucose and glycated hemoglobin are normal at the expense of very high insulin levels. Diabetes usually starts at puberty; in our series, the mean age of onset was 15.8±7.1 years27. Initially, they are controlled with oral drugs, needing high doses of insulin in a few years27. Arterial hypertension occurs in one-third of patients27.
There are some specific clinical features of each CGL type. Patients with type 1 still present mechanical adipose fat, especially in palms, soles, orbital, peri-articular regions31. In contrast, type 2 patients show an absence of metabolic and mechanical fatty tissues. Seipin is highly expressed in the brain and cerebellum and is also involved in the regulation of neural functions. More than half of type 2 patients have some cognitive impairment1, 8. Types 3 and 4 have preservation of mechanical and bone marrow fat, and type 4 has muscle weakness associated with high serum creatine kinase and spinal instability15.
There are also gender-specific clinical features. Polycystic ovaries and amenorrhea are common32. Menstrual cycles usually return to normal with the use of metreleptin, probably due to improvement in insulin sensitivity and restoration of LH pulsatility32. Type 2 men can have teratozoospermia due to the lack of seipin in germ cells33.
Hypertriglyceridemia occurs since the first years of life and can cause acute pancreatitis. HDL is usually lower than 30 mg/dL. Elevations of liver enzymes is also an early finding and come from the fat deposition in the liver. Progressive reductions in serum platelets suggest worsening of the liver disease and probable cirrhosis34.
As Cavin-1 is present in the muscle cells, patients with type 4 have mild muscle weakness and elevated creatine kinase15.
Life expectancy, mainly in type 2, is substantially decreased, with death not infrequently occurring before the age of 30 years (personal experience based on 20 patients who died in the last 19 years). The causes of death are related to diabetes (renal failure, sudden death), liver (cirrhosis, digestive bleeding) or infections.
Diagnosis and Treatment
The CGL diagnosis is based on clinical data: acromegaloid features, acanthosis nigricans, reduction of total body fat, muscular hypertrophy, and protrusion of the umbilical scar. Also, laboratory data can show diabetes with severe insulin resistance and hypertriglyceridemia. Imaging tests can help identify ectopic deposits of fat mainly in the liver and pancreas (hepatic steatosis with hepatomegaly and pancreatic steatosis). The DXA can confirm the low body fat and high bone density30.
The treatment of CGL consists of strict control of the diet with the decrease of the intake of fat, principally, triglycerides and foods with a high glycemic index to prevent and control comorbidities29. However, the ideal diet is a challenging goal to achieve because of the increased appetite and the severe restriction advocated. Physical activity should also be encouraged to improve control of comorbidities, except in those patients with contraindications such as severe cardiomyopathy29.
Regarding drug treatment, these patients can be treated with the usual medications for diabetes, hypertension, and dyslipidemia guidelines. The first choice for treatment of diabetes and insulin resistance is metformin, but usually, it is not enough. Unlike the treatment of partial lipodystrophy, thiazolidinediones should be used with caution29. Other oral antidiabetic agents are used, but they were not specifically studied in CGL patients. There are data in animals suggesting that the use of SGLT2 inhibitors (dapagliflozin) could have benefits preventing cardiomyopathy35; studies are needed to confirm this in humans. As the disease progresses and severe insulin resistance occurs, high daily doses of insulin are needed. The lack of subcutaneous adipose tissue is a problem in administering the high doses of insulin. More concentrated insulin (U-300 or U500) may be required36. These patients present severe dyslipidemia, mainly due to the increase of triglycerides and low HDL, and therefore, the use of fibrate is sometimes necessary to prevent acute pancreatitis. In addition, owing to the high cardiovascular risk of these patients, intervention with a statin should be considered, and the targets of LDL or non-HDL should be strict29.
Daily injections of metreleptin cause a significant decrease in appetite and bring benefits by lowering glycemia, triglyceridemia, and liver enzymes. It is notable, especially in children, the reduction of abdominal circumference, probably due to a reduction of hepatomegaly.
Conclusion
CGL is a rare and severe disease that can occur with diabetes (usually requiring high doses of insulin) and early death. The phenotype of the patient is quite characteristic, requiring, however, knowledge of the syndrome by the health professionals to make an early diagnosis. Metreleptin seems to be the only medication at the moment that can modify the natural history of the disease.
Conflict of interest: none.
References
- Nolis T. Exploring the pathophysiology behind the more common genetic and acquired lipodystrophies. Journal of human genetics. 2014 Jan; 59(1): 16-23.
- Agarwal AK, Arioglu E, De Almeida S, et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet. 2002 May; 31(1): 21-3.
- Magre J, Delepine M, Khallouf E, et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nature genetics. 2001 Aug; 28(4): 365-70.
- Patni N, Garg A. Congenital generalized lipodystrophies--new insights into metabolic dysfunction. Nature reviews Endocrinology. 2015 Sep; 11(9): 522-34.
- Garg A. Acquired and inherited lipodystrophies. The New England journal of medicine. 2004 Mar 18; 350(12): 1220-34.
- de Azevedo Medeiros LB, Candido Dantas VK, Craveiro Sarmento AS, et al. High prevalence of Berardinelli-Seip Congenital Lipodystrophy in Rio Grande do Norte State, Northeast Brazil. Diabetol Metab Syndr. 2017; 9: 80.
- Chiquette E, Oral EA, Garg A, et al. Estimating the prevalence of generalized and partial lipodystrophy: findings and challenges. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy. 2017: 375-83.
- Wee K, Yang W, Sugii S, et al. Towards a mechanistic understanding of lipodystrophy and seipin functions. Bioscience reports. 2014; 34(5).
- Dollet L, Magre J, Cariou B, et al. Function of seipin: new insights from Bscl2/seipin knockout mouse models. Biochimie. 2014 Jan; 96: 166-72.
- Sim MF, Dennis RJ, Aubry EM, et al. The human lipodystrophy protein seipin is an ER membrane adaptor for the adipogenic PA phosphatase lipin 1. Molecular metabolism. 2012; 2(1): 38-46.
- Sim MF, Talukder MM, Dennis RJ, et al. Analysis of naturally occurring mutations in the human lipodystrophy protein seipin reveals multiple potential pathogenic mechanisms. Diabetologia. 2013 Nov; 56(11): 2498-506.
- Payne VA, Grimsey N, Tuthill A, et al. The human lipodystrophy gene BSCL2/seipin may be essential for normal adipocyte differentiation. Diabetes. 2008 Aug; 57(8): 2055-60.
- Cohen AW, Hnasko R, Schubert W, et al. Role of caveolae and caveolins in health and disease. Physiological reviews. 2004 Oct; 84(4): 1341-79.
- Pilch PF, Liu L. Fat caves: caveolae, lipid trafficking and lipid metabolism in adipocytes. Trends in endocrinology and metabolism: TEM. 2011 Aug; 22(8): 318-24.
- Hayashi YK, Matsuda C, Ogawa M, et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009 Sep; 119(9): 2623-33.
- Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nature reviews Molecular cell biology. 2013 Feb; 14(2): 98-112.
- Le Lay S, Briand N, Blouin CM, et al. The lipoatrophic caveolin-1 deficient mouse model reveals autophagy in mature adipocytes. Autophagy. 2010 Aug; 6(6): 754-63.
- Liu L, Brown D, McKee M, et al. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell metabolism. 2008 Oct; 8(4): 310-7.
- Garg A, Wilson R, Barnes R, et al. A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. The Journal of clinical endocrinology and metabolism. 1999 Sep; 84(9): 3390-4.
- Vogel P, Read R, Hansen G, et al. Pathology of congenital generalized lipodystrophy in Agpat2-/- mice. Veterinary pathology. 2011 May; 48(3): 642-54.
- Cortes VA, Curtis DE, Sukumaran S, et al. Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell metabolism. 2009 Feb; 9(2): 165-76.
- Kim CA, Delepine M, Boutet E, et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008 Apr; 93(4): 1129-34.
- Rajab A, Straub V, McCann LJ, et al. Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. PLoS genetics. 2010 Mar 12; 6(3): e1000874.
- Payne F, Lim K, Girousse A, et al. Mutations disrupting the Kennedy phosphatidylcholine pathway in humans with congenital lipodystrophy and fatty liver disease. Proc Natl Acad Sci U S A. 2014 Jun 17; 111(24): 8901-6.
- Dyment DA, Gibson WT, Huang L, et al. Biallelic mutations at PPARG cause a congenital, generalized lipodystrophy similar to the Berardinelli-Seip syndrome. Eur J Med Genet. 2014 Sep; 57(9): 524-6.
- Garg A. Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. The Journal of clinical endocrinology and metabolism. 2011 Nov; 96(11): 3313-25.
- Lima JG, Nobrega LH, de Lima NN, et al. Clinical and laboratory data of a large series of patients with congenital generalized lipodystrophy. Diabetol Metab Syndr. 2016; 8: 23.
- Lima GJ, Lima NN, Oliveira CF, et al. Umbilical Hernia in Patients with Berardinelliseip Syndrome: Is it Really Hernia. J Clin Mol Endocrinol. 2015; 1(1): 3.
- Brown RJ, Araujo-Vilar D, Cheung PT, et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab. 2016 Dec; 101(12): 4500-11.
- Lima JG, Nobrega LH, Lima NN, et al. Bone Density in Patients With Berardinelli-Seip Congenital Lipodystrophy Is Higher in Trabecular Sites and in Type 2 Patients. J Clin Densitom. 2016 Nov 25.
- Simha V, Garg A. Phenotypic heterogeneity in body fat distribution in patients with congenital generalized lipodystrophy caused by mutations in the AGPAT2 or seipin genes. J Clin Endocrinol Metab. 2003 Nov; 88(11): 5433-7.
- Musso C, Cochran E, Javor E, et al. The long-term effect of recombinant methionyl human leptin therapy on hyperandrogenism and menstrual function in female and pituitary function in male and female hypoleptinemic lipodystrophic patients. Metabolism. 2005 Feb; 54(2): 255-63.
- Jiang M, Gao M, Wu C, et al. Lack of testicular seipin causes teratozoospermia syndrome in men. Proc Natl Acad Sci U S A. 2014 May 13; 111(19): 7054-9.
- Mitchell O, Feldman DM, Diakow M, et al. The pathophysiology of thrombocytopenia in chronic liver disease. Hepat Med. 2016; 8: 39-50.
- Joubert M, Jagu B, Montaigne D, et al. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Cardiomyopathy in a Diabetic Lipodystrophic Mouse Model. Diabetes. 2017 Apr; 66(4): 1030-40.
- Lima JG, Lima NN, Lima RLM, et al. Glargine U300 Insulin as a Better Option than Degludec U100 to Treat a Congenital Generalized Lipodystrophy Patient. Clin Diabetes Res. 2017; 1(1).