
DDHD domain-containing lipases: Targets for the treatment of rare diseases
Pradeep Kumar Yadav1,2 and Ram Rajasekharan1,2*
1Lipidomic Centre, Department of Lipid Science, CSIR-Central Food Technological Research Institute (CFTRI), Mysore 570020, Karnataka, India
2Academy of Scientific & Innovative Research, CSIR-CFTRI, Mysore, India
Abstract
The DDHD domain-containing lipases belong to the intracellular phospholipase A1 (iPLA1) family. Phospholipases have been implicated in the regulation of lipid metabolism, intracellular membrane trafficking, and signaling. In addition, phospholipases have been linked to the development of rare and neurodegenerative diseases. The rare and neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease have been focused on phospholipase A2. But there is a scarcity of literature on the role of PLA1 in rare and neurodegenerative diseases. Recently, in humans, mutation in DDHD1 and DDHD2 (iPLA1 members) has been identified as a cause of specific types of hereditary spastic paraplegia (HSP) termed as SPG28 and SPG54, respectively. Ddl1 (DDHD domain-containing lipase 1), a yeast homolog of human DDHD1/2, hydrolyzes cardiolipin (CL), phosphatidylethanolamine, and phosphatidylglycerol. Ddl1 has an important role in the mitochondrial phospholipids remodeling. Defects in phospholipids remodeling and mitochondrial functions have been implicated in the development of the Barth syndrome, HSPs, and other neurodegenerative disorders. Mutations in DDHD1 and DDHD2 produce DDL1-defective yeast strain like phenotypes (mitochondrial dysfunction and defects in lipid metabolism).
Therefore, the DDL1-defective yeast could be a good model system to understand hereditary spastic paraplegia.
DDHD domain
The DDHD domain was first identified as a long stretch of amino acids in the central part of the Nir/rdgB (N-terminal domain-interacting receptor/Drosophila retinal degeneration B) proteins1. This domain possesses four conserved amino acid residues (DDHD), which may play an important role in forming a metal-binding site. The name DDHD domain is based on these four conserved amino acid residues. This domain is also found in the C-terminal region of the phosphatidic acid (PA)-preferring phospholipase A1 (PA-PLA1)1. The recently identified lipid-metabolizing enzyme family, named as intracellular phospholipase A1(iPLA1) family also possesses the DDHD domain-containing proteins. It has been predicted that the DDHD domain may play a role in the phospholipid metabolism, organelle biogenesis, membrane trafficking, and signaling1. In humans, there are three iPLA1family members namely PA-PLA1/DDHD1, KIAA0725p/DDHD2 and p125/Sec23IP. The iPLA1 family members have also been reported in yeasts, nematodes, and plants2. The first iPLA1 member, PA-PLA1, was identified by Higgs and Glomset3. The DDHD14and DDHD25 have hydrolase activities, while the lipase activity of p125 is yet to be demonstrated. A study of the substrate specificity of the purified rat DDHD2 provides new insights into the enzymatic nature of the human DDHD26. The human DDHD1 and DDHD2 are the PA preferring lipases and play an important role in the intracellular membrane trafficking. PA has diverse functions in biological systems. It is a precursor for the biosynthesis of polar and non-polar lipids. It has structural functions in the biological membranes7. It has also been reported that PA has an important role in the lipid signaling8. Like iPLA1 family members, the DDHD domain is also conserved in some phosphatidylinositol transfer proteins9,10. Inoue et al. have shown that the DDHD domain is crucial for the PLA1 activity as well as in phosphatidylinositol phosphate (PIP) binding and oligomerization4. PIPs play crucial parts in the membrane trafficking11,12.
Role of phospholipases in rare diseases
Phospholipids are an important constituent of biological membranes. Several studies13-19 have shown that phospholipases play an important role in rare and neurodegenerative diseases. Phospholipases are enzymes that hydrolyze the phospholipids. Phospholipases have been implicated in the processes like regulation of lipid transport and metabolism, intracellular membrane trafficking, and vesicular transport13. Phospholipases are categorized in A, B, C and D groups according to their site of action. Most of the studies related to disease conditions have been focused on phospholipase A2 (PLA2)13-16. As different types of PLA2 have been identified in the central nervous system (CNS); therefore, the role of PLA2s in rare and neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease have been widely studied14-19. But the knowledge regarding the roles of different types of PLA2 is sketchy because of the complexity of the brain which possesses different types of cells. On the other hand, there is a scarcity of reports on the role of PLA1 in rare and neurodegenerative diseases. Recently, in humans, mutations in DDHD1 and DDHD2 (iPLA1 members) have been identified as a cause of specific types of hereditary spastic paraplegia termed as SPG2820 and SPG5421,22, respectively. Hereditary spastic paraplegia (HSP, also known as Strumpell-Lorrain disease) is a heterogeneous group of genetic neurodegenerative disorders, and these disorders are mainly characterized by slowly progressive spasticity (contraction) and weakness of the lower limbs. Many types of HSP have been discovered and are numbered according to the order of their discovery [spastic paraplegia (SPG) 1-72]. Affected individuals are clinically grouped according to the absence (uncomplicated or pure HSP) or presence (complicated or complex HSP) of additional phenotypes23,24. By modes of inheritance, HSPs are grouped as autosomal dominant, autosomal recessive, and X-linked recessive HSPs23,24.
DDHD domain-containing lipase in Saccharomyces cerevisiae
We25 have characterized an unannotated Saccharomyces cerevisiae open reading frame, YOR022C as a Ddl1 (DDHD domain-containing lipase 1). Pairwise sequence alignment showed that yeast Ddl1 has ~33% and ~34% similarities with human DDHD1 and DDHD2, respectively. The Ddl1 is a novel mitochondrial phospholipase of the iPLA1 family (Figure 1) that hydrolyzes important mitochondrial phospholipids, and it has roles in cardiolipin (CL), phosphatidylethanolamine (PE), and phosphatidylglycerol (PG) remodeling. The deletion of the DDL1 gene caused an increase in molecular species with saturated fatty acids while DDL1 overexpression caused an increase in the molecular species with unsaturated fatty acids. In addition, we have shown that Ddl1 has roles in CL degradation. Our findings showed that Ddl1 is a CL-preferring lipase; therefore deletion of the DDL1 gene caused an increase in the CL content while overexpression of the DDL1 gene caused a decrease in the cellular CL content. Also, the CL degradation was affected in the DDL1 deleted cells (monolysocardiolipin to dilysocardiolipin conversion was affected by the deletion of the DDL1 gene), leading to the CL accumulation. On the other hand, the DDL1 overexpression increased the CL degradation leading to reduced CL content. Therefore, optimal regulation and activity of Ddl1 are required for the proper CL metabolism. As CL is the signature phospholipid species of the mitochondria; misregulation of Ddl1 causes mitochondrial dysfunction in yeast25.
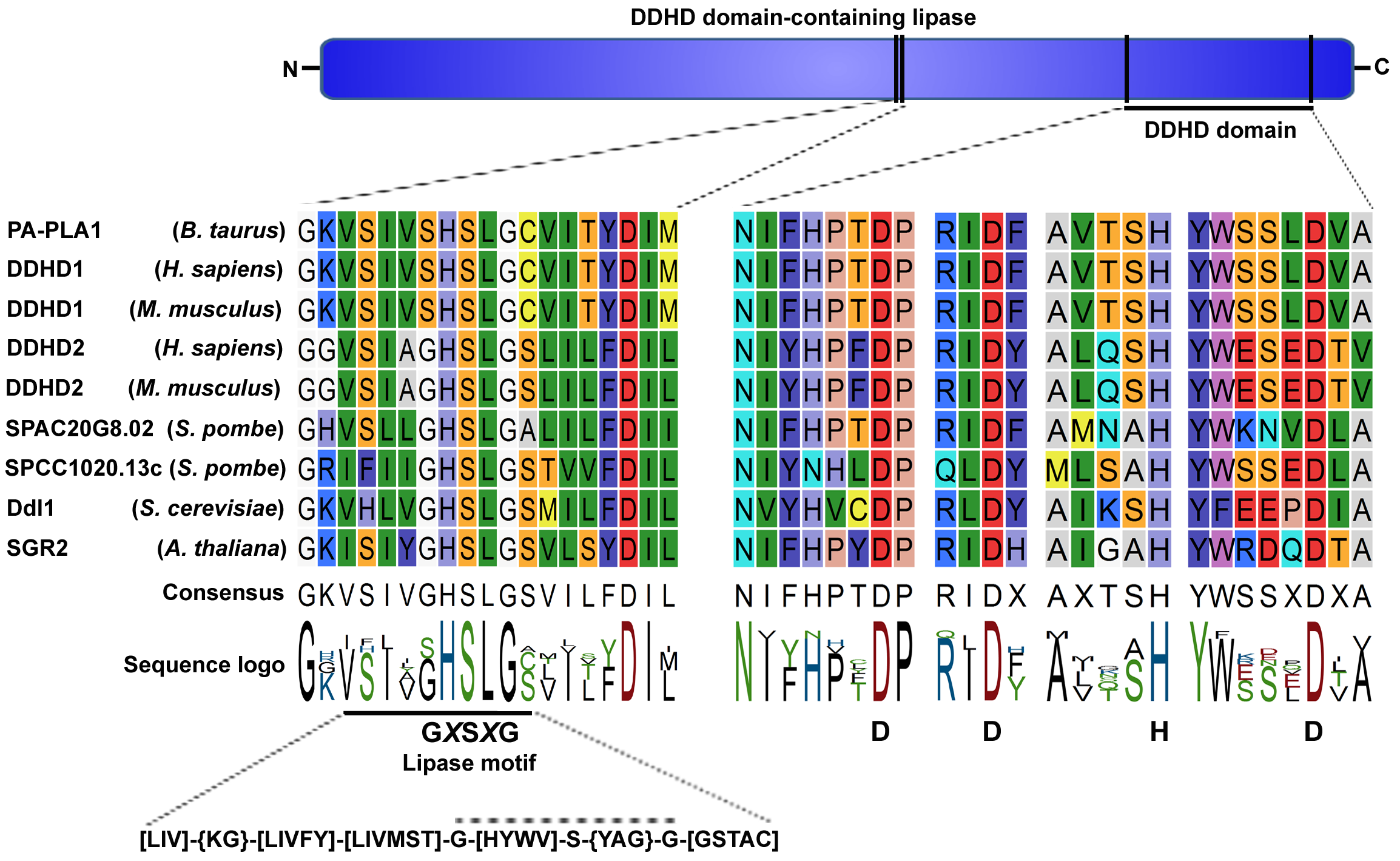
Figure 1: Ddl1 and its homologs.
A schematic diagram of the DDHD domain-containing lipase and multiple sequence alignment of two conserved regions: the lipase motif (GXSXG) and DDHD domain. A list of homologs of the iPLA1 family is available on the UniProtKB database. An analysis of the sequences in the PROSITE database showed that the lipase motif is present inside the PROSITE domain PS00120 consensus sequence [LIV]-{KG}-[LIVFY]-[LIVMST]-G-[HYWV]-S-{YAG}-G-[GSTAC]. In some family members, the lipase consensus sequence (GXSXG) was replaced by the SXSXG sequence. In Ddl1, the lipase motif spanned from the 499th to 503rd amino acids, while the DDHD domain spanned from the 519th to 700th amino acids.
Role of mitochondria in rare diseases
The mitochondria are vital organelles of the cell. Important metabolic reactions and the regulation of some signaling cascades occur in the mitochondria26. The mitochondria are sites of synthesis of major non-bilayer-forming phospholipids PE and CL27,28. CL and PE play an important role in the mitochondrial fusion29. CL is required for the proper mitochondrial inner membrane potential Δψ and in protein import into the mitochondria30. CL and mitochondrial PE have overlapping functions, and they can compensate for the loss of the other31. Defective mitochondrial phospholipids remodeling has been identified as a major cause of Barth syndrome32,33. We25 have proposed that CL is remodeled through a deacylation-transacylation cycle, in which the acyl specificity of CL is achieved by extensive remodeling through PLA1 (Ddl1), PLA2 (Cld1), and transacylase (Taz1) activities. A deacylation (by Ddl1)-reacylation (by CoA-dependent acyltransferase, Ale1) cycle was proposed for the PE and PG remodeling in yeast (Figure 2).
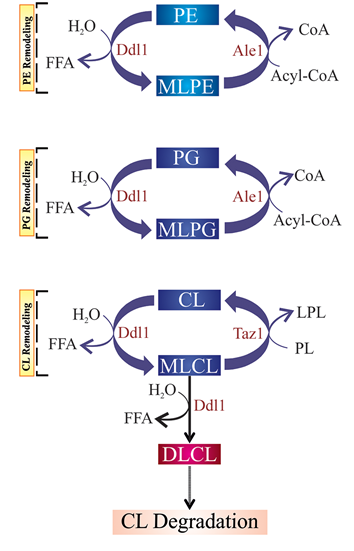
Figure 2: A schematic model of the proposed roles of the DDL1 gene in yeast
We propose that Ddl1 plays an important role in phosphatidylethanolamine (PE), phosphatidylglycerol (PG) and cardiolipin (CL) remodeling; as well as CL degradation. Ddl1 overexpression enhances the degradation of the mitochondrial membrane lipids, causing mitochondrial dysfunction in yeast. MLPE, monolysophosphatidylethanolamine; MLPG, monolysophosphatidylglycerol; MLCL, monolysocardiolipin; DLCL, dilysocardiolipin; and LPL, lysophospholipid.
It has been shown that human DDHD1 is partially localized to the mitochondria20,34 and mutations in DDHD1 gene produce mitochondrial dysfunction and defects in lipid metabolism. The DDHD2 protein has been reported to locate in cis-Golgi and endoplasmic reticulum (ER)-Golgi intermediate compartment, besides role of DDHD2 in the membrane trafficking and vesicle fusion has been proposed4. Defects in the intracellular membrane trafficking, mitochondrial morphology and functions, and lipid metabolism are key pathophysiological features of HSPs20,35. The neurodegenerative disorders, such as Alzheimer’s disease, and Huntington’s disease have also been associated with the defects in the intracellular membrane trafficking, mitochondrial morphology, and functions36. Alteration in the mitochondrial-membrane lipid composition has already been shown to cause mitochondrial dysfunction which in turn triggers secondary cellular dysfunctions37-39. The increased reactive oxygen species (ROS) production was observed in SPG28 and SPG49 cells, and it was hypothesized that the increased ROS production could cause neurodegeneration40.
A Recent study has linked alterations in CL profile to an early development of the age-related neurodegenerative disorders such as Alzheimer’s disease41. Human HSD10 is a multifunctional enzyme which is found in the brain and cerebral spinal fluid and has been associated with the mitochondrial disease. In Alzheimer’s disease patients, the HSD10 expression was found elevated42. Recently, a study showed that the HSD10 protein has CL-specific phospholipase C-like enzyme activity43.
Cures and future perspectives
In humans, many rare genetic diseases are associated with mutations in poorly characterized genes. Determination of biochemical functions of these genes is critical for understanding and formulating potential cures for these rare genetic diseases. The probable implication of cardiolipin and mitochondria in the pathophysiology of neurodegenerative disorders could help in the development of therapeutic strategies focused on the mitochondrial morphology and functions.
Polyunsaturated fatty acids (PUFAs) are enriched in the phospholipids of CNS membranes44. Metabolism of PUFA is strictly controlled by PLA2 and acyltransferases through “deacylation-reacylation cycle” which is an energy-dependent process involving coenzyme A (CoA) and ATP45-47. We25 have shown that Ddl1 (PLA1), the yeast homolog of human DDHD1 and DDHD2 proteins, plays an important role in the mitochondrial phospholipids remodeling. Therefore, it will be interesting to study the role of DDHD1 and DDHD2 proteins in phospholipids remodeling.
There are several synthetic and phytochemical based inhibitors of PLA2 available and which could be used for the treatment of neurological disorders48. Ginkgo biloba and Centella asiatica extracts have been used for the treatment of neurological disorders in the cell culture and animal model systems49,50. The neurological disorders wherein the PLA2 expression is elevated, PLA2inhibitors could be used for the treatment. Likewise, identification of PLA1 inhibitors would provide a base for the innovative development of therapeutic strategies focused on the PLA1 activities.
Cardiolipin is always at the risk of oxidation as it is localized to the inner mitochondrial membrane. When cardiolipin becomes oxidized, it induces apoptosis and could trigger diseases such as Alzheimer's and Parkinson's. Researchers have suggested that HSD10 protein has CL-specific phospholipase C-like enzyme activity, and it prevents neurodegeneration by removing oxidized cardiolipin43. In humans, mutations in the DDHD1 and DDHD2 genes cause specific types of hereditary spastic paraplegia, and the yeast DDL1-defective strain exhibits the similar phenotypes of hereditary spastic paraplegia (mitochondrial dysfunction and defects in lipid metabolism). Yeast Ddl1 prefers CL as substrates. It has also been shown that DDHD1 is partially localized to the mitochondria20,34. Therefore, it will be interesting to explore the role of DDHD1 in CL metabolism. In addition, the DDL1-defective yeast strain could be a suitable model system to understand hereditary spastic paraplegia.
Acknowledgements
This study was supported by the Council of Scientific and Industrial Research (CSIR), New Delhi, under the 12th five-year plan project LIPIC. P.K.Y. was supported by a fellowship from CSIR, New Delhi. The corresponding author is a recipient of the JC Bose national fellowship. The authors declare that they have no conflicts of interest.
References
- Lev S. The role of the Nir/rdgB protein family in membrane trafficking and cytoskeleton remodeling. Exp Cell Res. 2004; 297: 1-10.
- Tani K, Kogure T, Inoue H. The intracellular phospholipase A1 protein family. Biomol Concepts. 2012; 3: 471-478.
- Higgs HN, Glomset JA. Identification of a phosphatidic acid-preferring phospholipase A1 from bovine brain and testis. Proc Natl Acad Sci U S A. 1994; 91: 9574-9578.
- Inoue H, Baba T, Sato S, Ohtsuki R, Takemori A, Watanabe T, et al. Roles of SAM and DDHD domains in mammalian intracellular phospholipase A1 KIAA0725p. Biochim Biophys Acta. 2012; 1823: 930-939.
- Inloes JM, Hsu KL. The hereditary spastic paraplegia-related enzyme DDHD2 is a principal brain triglyceride lipase. Proc Natl Acad Sci U S A. 2014; 111: 14924-14929.
- Araki M, Ohshima N, Aso C, Konishi A, Obinata H, Tatei K, et al. Enzymatic characterization of recombinant rat DDHD2: a soluble diacylglycerol lipase. J Biochem. 2016.
- Kooijman EE, Chupin V, de Kruijff B, Burger KN. Modulation of membrane curvature by phosphatidic acid and lysophosphatidic acid. Traffic 2003; 4: 162-174.
- Wang X, Devaiah SP, Zhang W, Welti, R. Signaling functions of phosphatidic acid. Prog Lipid Res. 2006; 45: 250-278.
- Cockcroft S, Carvou N. Biochemical and biological functions of class I phosphatidylinositol transfer proteins. Biochim Biophys Acta. 2007; 1771: 677-691.
- Lev S. Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat Rev Mol Cell Biol. 2010; 11: 739-750.
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006; 443: 651-657.
- Vicinanza M, D'Angelo G, Di Campli A, De Matteis MA. Function and dysfunction of the PI system in membrane trafficking. EMBO J. 2008; 27: 2457-2470.
- Brown WJ, Chambers K, Doody A. Phospholipase A2 (PLA2) enzymes in membrane trafficking: mediators of membrane shape and function. Traffic. 2003; 4: 214-221.
- Phillis JW, O'Regan MH. A potentially critical role of phospholipases in central nervous system ischemic, traumatic, and neurodegenerative disorders. Brain Res Rev. 2004; 44: 13-47.
- Farooqui AA, Horrocks LA, Farooqui T. Glycerophospholipids in brain: their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem Phys Lipids. 2000; 106: 1-29.
- Sun GY, Xu J, Jensen MD, Simonyi A. Phospholipase A2 in the central nervous system: implications for neurodegenerative diseases. J Lipid Res. 2004; 45: 205-213.
- Cummings BS, McHowat J, Schnellmann RG. Phospholipase A(2)s in cell injury and death. J Pharmacol Exp Ther. 2000; 294: 793-799.
- Murakami M, Nakatani Y, Atsumi G, Inoue K, Kudo I. Regulatory functions of phospholipase A2. Crit Rev Immunol. 1997; 17: 225-283.
- Gentile MT, Reccia MG, Sorrentino PP, Vitale E, Sorrentino G, Puca AA, et al. Role of cytosolic calcium-dependent phospholipase A2 in Alzheimer's disease pathogenesis. Mol Neurobiol. 2012; 45: 596-604.
- Tesson C, Nawara M, Salih MA, Rossignol R, Zaki MS, Al Balwi M, et al. Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am J Hum Genet. 2012; 91: 1051-1064.
- Gonzalez M, Nampoothiri S, Kornblum C, Oteyza AC, Walter J, Konidari I, et al. Mutations in phospholipase DDHD2 cause autosomal recessive hereditary spastic paraplegia (SPG54). Eur J Hum Genet. 2013; 21: 1214-1218.
- Schuurs-Hoeijmakers JH, Geraghty MT, Kamsteeg EJ, Ben-Salem S, de Bot ST, Nijhof B, et al. Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am J Hum Genet. 2012; 91: 1073-1081.
- Harding AE. Hereditary spastic paraplegias. Semin Neurol. 1993; 13; 333-336.
- Fink JK. Advances in the hereditary spastic paraplegias. Exp. Neurol. 2003; 184 Suppl 1: S106-110.
- Yadav PK, Rajasekharan R. Misregulation of a DDHD domain-containing lipase causes mitochondrial dysfunction in yeast. J Biol Chem. 2016; 291:18562-18581.
- Dimmer KS, Scorrano L. (De)constructing mitochondria: what for? Physiology (Bethesda, Md.). 2006; 21: 233-241.
- Schuiki I, Daum G. Phosphatidylserine decarboxylases, key enzymes of lipid metabolism. IUBMB life. 2009; 61: 151-162.
- Claypool SM, Koehler CM. The complexity of cardiolipin in health and disease. Trends Biochem Sci. 2012; 37: 32-41.
- Joshi AS, Thompson MN, Fei N, Huttemann M, Greenberg ML. Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J Biol Chem. 2012; 287: 17589-17597.
- Jiang F, Ryan MT, Schlame M, Zhao M, Gu Z, Klingenberg M, et al. Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J Biol Chem. 2000; 275: 22387-22394.
- Gohil VM, Thompson MN, Greenberg ML. Synthetic lethal interaction of the mitochondrial phosphatidylethanolamine and cardiolipin biosynthetic pathways in Saccharomyces cerevisiae. J Biol Chem. 2005; 280: 35410-35416.
- Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJ, et al. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun. 2000; 279: 378-382.
- Schlame M, Kelley RI, Feigenbaum A, Towbin JA, Heerdt PM, Schieble T, et al. Phospholipid abnormalities in children with Barth syndrome. J Am Coll Cardiol. 2003; 42: 1994-1999.
- Yamashita A, Kumazawa T, Koga H, Suzuki N, Oka S, Sugiura T. Generation of lysophosphatidylinositol by DDHD domain containing 1 (DDHD1): Possible involvement of phospholipase D/phosphatidic acid in the activation of DDHD1. Biochim Biophys Acta. 2010; 1801: 711–720.
- Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci. 2012; 35: 25-47.
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006; 443; 787-795.
- Steenbergen R, Nanowski TS, Beigneux A, Kulinski A, Young SG, Vance JE. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J Biol Chem. 2005; 280: 40032-40040.
- Brandner K, Mick DU, Frazier AE, Taylor RD, Meisinger C, Rehling P. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth Syndrome. Mol Biol Cell. 2005; 16: 5202-5214.
- Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell Biol. 2006; 8: 1255-1262.
- Gucuyener K, Pinarli FG, Erbas D, Hasanoglu A, Serdaroglu A, Topaloglu H. Is oxidative damage in operation in patients with hereditary spastic paraparesis? Brain Dev. 2010; 32: 130-136.
- Monteiro-Cardoso VF, Oliveira MM, Melo T, Domingues MR, Moreira PI, Ferreiro E, et al. Cardiolipin profile changes are associated to the early synaptic mitochondrial dysfunction in Alzheimer's disease. J Alzheimers Dis. 2015; 43: 1375-1392.
- Yang SY, He XY, Isaacs C, Dobkin C, Miller D, Philipp M. Roles of 17beta-hydroxysteroid dehydrogenase type 10 in neurodegenerative disorders. J. Steroid Biochem. Mol Biol. 2014; 143: 460-472.
- Boynton TO, Shimkets LJ. Myxococcus CsgA, Drosophila Sniffer, and human HSD10 are cardiolipin phospholipases. Genes Dev. 2015; 29: 1903-1914.
- Sun GY, Horrocks LA. The acyl and alk-1-enyl groups of the major phosphoglycerides from ox brain myelin and mouse brain microsomal, mitochondrial and myelin fractions. Lipids. 1970; 5: 1006-1012.
- Farooqui A A, Horrocks L A, Farooqui T. Deacylation and reacylation of neural membrane glycerophospholipids. J Mol Neurosci. 2000; 14: 123-135.
- Sun GY, MacQuarrie RA. Deacylation-reacylation of arachidonoyl groups in cerebral phospholipids. Ann N Y Acad Sci. 1989; 559: 37-55.
- Corbin DR, Sun GY. Characterization of the enzymic transfer of arachidonoyl groups to 1-acyl-phosphoglycerides in mouse synaptosome fraction. J Neurochem. 1978; 30: 77-82.
- Ong WY, Farooqui T, Kokotos G, Farooqui AA. Synthetic and natural inhibitors of phospholipases A2: their importance for understanding and treatment of neurological disorders. ACS Chem Neurosci. 2015; 6: 814-831.
- Zhao Z, Liu N, Huang J, Lu PH, Xu XM. Inhibition of cPLA2 activation by Ginkgo biloba extract protects spinal cord neurons from glutamate excitotoxicity and oxidative stress-induced cell death. J Neurochem. 2011; 116: 1057-1065.
- Barbosa NR, Pittella F, Gattaz WF. Centella asiatica water extract inhibits iPLA2 and cPLA2 activities in rat cerebellum. Phytomedicine. 2008; 15: 896-900.