
Ralf‐Dieter Hilgers1*, FranzKönig2, Geert Molenberghs3 and Stephen Senn4
1Department of Medical Statistics, RWTH Universit Aachen, Pauwelstr 30, D‐ 52074 Aachen, Germany
2Center for Medical Statistics, Informatics and Intelligent Systems, Medical University of Vienna, Vienna, Austria
3I‐BioStat, Universiteit Hasselt, B‐3590 Diepenbeek, Belgium
4Competence Centre for Methodology and Statistics, Luxembourg Institute of Health, L‐1445 Strassen, Luxembourg
The mostly unmet need as well as the pressure to show efficacy of new therapies to treat rare diseases contrasts with the limited possibility to use traditional statistical methods to design and analyse clinical trials in this setting. Within this paper, we will refer to the current state of design and analysis methods, as well as practical conditions to be considered when conducting a clinical trial for rare diseases. We will embed the research of the IDeAl project within this setting and give some first recommendations to improve the methodology for clinical trials in rare diseases.
Common to the definition of rare diseases is the relative frequency of the number of affected patients in the parent population. Thus, a disease is considered as rare if fewer than 1 out of 2000 in the EU, 1500 in the US and 2500 in Japan are affected. The number of rare diseases is estimated to be 6‐80001. However, for some rare diseases it is not difficult to prove the efficacy of a new treatment, because the population size is relatively large. For instance the incidence of Friedreich Ataxia in the general population is roughly 1 of 50,000 resulting in 10,000 new patients in the EU. However the majority of rare diseases are less frequent2. Most of the diseases cannot be treated adequately. Consequently the International Rare Disease Research Consortium has stated the objective to make the diagnosis of as many rare diseases as possible and to contribute to the development of 200 new rare disease treatments by 20203.
The problem with drug approval in rare diseases is among others related to the limited evidence resulting from clinical trials in small populations. The methodological framework on clinical trials in small populations from the regulator perspective is described in the EU by the EMA guidance4 and in the US by the draft guidance on rare disease5. The framework covers aspects like levels of evidence, pharmacological considerations, choice of endpoints and control group as well as methodological and statistical considerations. Especially with respect to paediatric trials similar problems occur and the recent EMA concept paper on extrapolation of efficacy and safety in medicine development6, proposes a general framework for adopting a scientific proof in a larger population, usually adults, to a much smaller population, usually children. Obviously, a similar problem is present for diseases with prevalences that vary by continent. For example, IgA nephropathy is rather rare in the EU but more frequent in Asia and Africa, so that one might be interested to adopt proofs of efficacy from the larger populations to the EU population.
Recognizing that the performance of methodological tools appears to be rather unsatisfactory or not well understood with regard to validity, when the sample size deteriorates, in 2012 the EU announced the call ‘New methodologies for clinical trials for small population groups’ within the FP7 health innovation framework. The designated objective is to develop new or improved statistical methodologies for clinical trials aiming at the efficient assessment of the safety and/or efficacy of a treatment for small population groups in particular for rare diseases or personalised (stratified or individualised) medicine. The expected impact is to reduce design costs and deliver efficient clinical trials deriving reliable results from trials in small population groups.
In this paper we report the views of the IDeAl research consortium (www.ideal.rwth‐ aachen.de) concerning potential improvement of the design and analysis of clinical trials for small population with special interest in rare diseases. To set the scene, we start with a report about important aspects mentioned in selected recently published papers. We are not providing a systematic review about these aspects. Realizing that statistical methods for clinical trials in small populations are rather specific to the disease under consideration, a systematic review is infeasible. In the final section, we will comment on these methods and give some recommendations.
In what follows we will describe the most important practical aspects that affect the development of new methodologies for clinical trials in small population groups. Then we will give an overview on trial designs and analysis methods and end up with some more specific aspects.
There is a growing pressure for (orphan) drug approvals to treat rare diseases from patients, health care bodies, governments etc. Based on the special situation with rare diseases there are some specific challenges. First, many rare diseases affect children with an unmet need for a therapy, there is a tendency to relax well established standards for treatment evaluation to bring new treatments faster to the patients. Second, since many rare diseases are supposed to be heterogeneous, it can be argued, that it is difficult to obtain a clinically relevant study population. Clinical parameters are difficult to define because many rare diseases are poorly characterized and under‐researched. This in particular refers to the difficulty of estimating the expected effect size of a therapy and to decide on the most appropriate duration of the study because of the limited knowledge about the natural cause of the disease7. Logistical problems are related to small number of patients and specialist centres7. The increasing number of “first in class” drugs8 causes reservation of some stakeholders based on limited knowledge of a new and unique mechanism for treatment of the disease. On the other hand, the estimated number of rare disease of 6000 to 8000 is rapidly increasing, because improved diagnostics lead to more segmented diseases1.
Recruitment to a rare disease clinical trial is frequently mentioned as the major problem. In large or common clinical trials multicentre layouts, whether international or not, are often recommended to overcome recruitment problems. However, there are several practical challenges with international multicentre trials which are special with rare diseases like consensus among clinical experts and regulatory agencies about fundamental questions like uncertainties about the correct diagnosis in small centres and about consistency across centres, measurement of endpoints in cross cultural studies, etc.9 Although it is mentioned that the collaboration between sponsor, academia and regulatory agencies is the prime determinant of a trial’s success9, the patient perspective should not be underestimated. All of these groups should be involved in the early stage of the protocol development process, as they often are the best ones to define the relevant clinical endpoints, identify specialised centres and disseminate information about the study to the patients7. Furthermore, with many rare diseases well‐organised patient advocacy groups under a European umbrella participate in rare disease specific registries. This information is, however, currently rarely used for proof of efficacy. Further recruitment is prolonged because of geographically wide spread distribution and small number of patients within centres. New recruitment strategies different from the “if you build it they will come” idea cover aspects like a more active search of patients, most notably telephone reminders, open–trial designs and opt‐out strategies as well financial incentives are recommended10. Furthermore information on a patient’s home‐based care using modern methods of data capture, including electronic devices for continuous monitoring, such as iPads, may be helpful in overcoming recruitment problems11.
There is a considerable amount of information in rare diseases from observational studies. Although the number of 651 registries listed by the Orphanet report2 is small compared to the estimated 6000 to 8000 diseases, registries may serve as an important tool of information to study the natural history of a disease as well as to improve designing a clinical trial from various perspectives. These registries provide relatively large representative cohorts. Some authors recommend putting more emphasis on observational studies, like self‐ controlled observational studies, case‐control studies and prospective inception cohort studies12. Registries can also serve as a basis for a randomized controlled trial13. Caution is necessary because it is argued that the high heterogeneity in phenotypic expression of many rare diseases may hinder optimal natural history and outcome studies based on registries14. However the same argument is frequently applied within the context of randomized clinical trials, e.g. by defining a suitable clinical population. Case‐control studies are useful for studying rare disease15. Applying this study type to rare disease registries matching techniques are found to minimize bias14.
Clinical trial designs with orphan drug approvals compared to non‐orphan drugs differ in various aspects. Most authors found that the pivotal studies for orphan drug approvals were more likely to be smaller, do not use placebo control, and use nonrandomized, un‐blinded trial design, e.g. single arm design and surrogate endpoints to assess efficacy16-18. On the other hand, a survey in ClinicalTrials.gov shows, that Bayesian methods and adaptive randomisation, although recommended in the guideline are not used16.
To give some figure about “what is a small trial” one can refer to the 63 orphan drug approvals in the EU from 2000 to 2010. Here 22 of 38 randomised controlled trials showing a total sample size below 5019. Given these figures, which suggest that the clinical trials are often small, one may question the relevance of the long run properties of randomization, the most important design technique to avoid bias in clinical trials. To address this the IDeAl consortium has developed methods to evaluate the impact of supposed bias type on the test decision, developed a selection bias corrected test and developed a software to evaluate and conduct a randomisation procedure20-22. The next step is to publish a framework for choosing the best practice randomization procedure for a small clinical trial as well as the corresponding randomization‐based inference. This investigation should become a standard procedure, when designing a clinical trial, in particular a small one.
On the other hand, some argue to substitute the control group in a clinical trial by historical controls which, however, have been assessed as a non‐satisfactory solution23. Using external controls in clinical trials involves careful analysis and skilful adjustment24.
Adaptive designs have been proposed as a means of gaining efficiency in studying rare diseases25,26,12. A recent review27 showed that about 59% of adaptive designs evaluated by the scientific advice working party of the European Medicines Agency related to trials in rare diseases and about 36% applied for orphan designation. The most attractive adaptation is sample size reassessment based on interim data. Adaptive designs use accumulating data of an ongoing trial to decide how to modify design aspects without undermining the validity and integrity of the trial. Based on interim data a trial may be stopped for efficacy or futility like in group sequential designs. Especially adaptive seamless designs28,29 seem to be very attractive for rare diseases, where sample sizes lack to conduct a series of independent phase II and phase III trials. Adaptive seamless designs are the combination of a clinical phase II study (focusing on treatment selection, for example) with a phase III study (confirmatory testing of treatments) allowing treatment selection and sample size re‐assessment at a pre‐defined interim analysis. The IDeAl consortium showed that there is a huge inflation in the type 1 error rate if treatment selection and sample size reassessment are not addressed adequately in the design and analysis of such seamless trials30. There are also some caveats when the endpoint is survival31. The IDeAl consortium proposes to use modelling techniques like MCPMod32 within the framework of adaptive seamless designs to address these objectives in an efficient way. Furthermore adaptive designs have been proposed for population or endpoint selection, adaptive dose finding, e.g., with continual reassessment methods, or adapting the allocation process using either covariates or early observable outcomes, i.e. response adaptive randomization. Most techniques have extensively been evaluated with respect to large sample theory; but their validity has rarely been explored for small clinical trials. For instance, the attractive property of response adaptive randomization e.g. play the winner, drop the looser, Klein’s urn design etc. to allocate more patients to the more effective treatment have not been evaluated with respect to small samples. So from the practical point of view it has to be evaluated how many patients must be included in the trial to gain efficiency. The IDeAl consortium evaluates the gain in efficiency of response adaptive randomization techniques compared to parallel group designs and adaptive designs with a single interim analysis with respect to small samples.
Within patient designs including repeated measures, crossover, Latin Square, stepped wedge and n‐of‐1 design are expected to be applicable and more efficient than the traditional parallel group designs. Taking into account that many rare diseases are chronic conditions within‐ patient designs like crossover designs are promising12. However there are various limitations, e.g. carryover effects, which meant that caution is required in applying the design. Nevertheless, where applicable, such within‐patient studies can, not only bring considerable gains in efficiency, but also permit careful study of individual response to treatment, and one of the threads of the IDeAL project has been devoted to studying their potential.
Various claims have been made about superior designs from cross‐over trials allowing for carry‐over effects. These claims should be treated with caution since the models involved are not very realistic. The example of a non‐linear dose response for a dose‐finding trial arranged in a Williams square is developed to show that if carry‐over is present to any appreciable degree the usual statistical models provide no guaranteed protection against its effects. It is concluded that the most reasonably defended assumption about carry‐over effects is that no important carry‐over has taken place and that, where this assumption cannot be defended, statistical models provide no satisfactory substitute for it33. Thus if the researcher can not exclude the existence of carryover effects a crossover design should not be considered. Similarly the selection of the stepped wedge design34, recommended because of ethical reasons and acceptance by the patients should be carefully considered. Here the variance of the simplest ABB/AAB stepped wedge design is 4 times the variance of the optimal crossover design ABB/BAA. Both cases might show the efficiency can be gained in practice and research should be aware of these results.
There are a lot of other designs with unknown efficiency with respect to small sample size like randomized withdrawal design, randomized placebo phase design, early escape design, delayed start design, re‐randomized designs35, dog leg design36, platform trials, basket designs and so on. Care should be given to uncritical application of such designs without any evaluation about the intended benefit.
Various recommendations concern the analysis of small clinical trials. The recommendation cover aspects to adjust the risk of erroneous decisions from clinical trials by carrying out low power clinical trials24 as well as accept greater type 1 error rate (10% onesided = 20% twosided)37. The suggestions may be contrasted to the ethical implications38 and the reliability of the study results39. Although the pressure in unmet clinical need scenarios in particular in rare diseases is high suggesting somewhat relaxed benefit risk assessment in particular by patients, the IDeAl consortium contrast these aspects with decision‐theoretic arguments. The point is not only whether or not to relax the standard margins, but also to give a scientific basis as to how much relaxing is reasonable. Including all stakeholder perspectives, i.e. patients, regulators, industry, reimbursers and academics, for a tailored decision about the most efficient design and analysis approach, would be the scientific solution to the ideas mentioned above.
Some advocate as analytical strategies including exact procedure, hierarchical models, including modelling the pattern of “missingness”25. With respect to exact procedures, a first approach could be to think about nonparametric tests, like permutation tests40. The IDeAl consortium realizes that the usual approaches with population based inference is hard to justify in a limited population. The uncertainty of this approach is answered by considering randomization based inference. Within this context, the randomization based inference within hierarchical models is investigated as well. Further, the question about the evaluation of the natural cause of a disease is answered by the consortium with methods to analyse reliability in longitudinal small data sets.
A lot of discussions focus on the choice of endpoints. It seems to be specific to rare diseases, to switch to more patient relevant endpoints. There are initiatives to define and find relevant endpoints like COMET13,41 as well as disease specific initiatives, e.g. [www.treat‐ nmd.eu/research/outcome‐measures/about/]. The effect of multiple endpoints as well as relevant effects in subscales need further investigation.
There is a broad consensus, that some benefit in the drug development program can be gained by surrogate endpoints in particular for rare diseases24,40,7. The problem with surrogate endpoints is, they may lack of clinical relevance, may not allow to measure the clinical benefit against adverse effects and their reliability is questionable. The IDeAl consortium develops a framework for validation of surrogate endpoints based on linear mixed‐effects and other hierarchical models, taking into account that there is less information available in the data than is usually the case. They also developed a framework for establishing reliability. This poses specific computational challenges.
Bayesian ideas are assessed to be helpful in various areas for therapy evaluation in rare diseases. There are two obvious purposes for which Bayesian methods can in principle be useful. The first is where different stakeholders have different utilities or prior beliefs. Incorporating these into a formal Bayesian analysis is a way of examining to what extent these impinge of potential decision‐making as a way of resolving possible conflicts. This was a key feature of the ‘Bayesian Approach to Randomised Trials, proposed over 20 years ago42. As already mentioned earlier, clinical decision making in rare cancers involving all stakeholders24 is one aspect, where such Bayesian ideas are used.
The second use of Bayesian methods is as a technique for combining information from disparate sources, for example not only randomised clinical trials, but also registries and observational studies generally. For example, using Bayesian approaches as an analytical strategy25 [e.g. use hierarchical Bayesian meta‐analysis model to analyse combined results from n‐of‐1 trials13, designing clinical trials24,26 and to quantify resulting levels of information, and to incorporate external information37 is recommended. People increasingly want to be informed, empowered and engaged with their medical management, providing better information to participants. This can be realized by patient centeredness in the design of clinical trials and use of Bayesian adaptive trials to adjust for changes in clinical practice in a prespecified manner26. The IDeAl consortium uses Bayesian ideas to design clinical trials adaptively, for extrapolation purposes43 and for clinical decision making.
There is considerable scope for improving drug development in rare diseases by using the promise of integrative mathematical analysis applied to pharmacokinetic‐pharmacodynamic models for selected drug candidates to optimize Phase III trial designs44. This implies the need for pharmacokinetic‐pharmacodynamics models as well as animal models for rare diseases. This is the point where “in silico” clinical trials start and may result in knowledge about the variability. The IDeAl consortium put emphasis on these questions by exploring non‐ linear mixed effects models as an important statistical tool to allow these aspects become available to better design small clinical trials. Further statistical methods for identification of interactions between the treatment and the genetic background are necessary for selecting groups of patients for personalized therapies as well as for identification of proper blocks of patients for randomized trials.
Another aspect which may be used to recommend new treatments for rare diseases is to use already existing knowledge so to avoid unnecessary clinical trials. This means to look for a drug, which is already in clinical use for a more common disease in case it is supposed to be efficacious within the rare disease as well45. This problem can be identified as an extrapolation. Here the dose‐response information is mentioned as a particular important topic in the analysis of small population groups6 because by the transfer of knowledge from larger to smaller populations it is possible to obtain information in cases where much data is available and thus to avoid unnecessary studies. This is helpful e.g. if diseases are rare in the EU, like IgA nephropathy, but more common in other continents, like Africa or Asia. To apply therapeutic options, evaluated in larger populations could be one way to overcome the troubles with the conduct of a clinical trial in small population groups.
We have referred to various actual aspects of statistical methodologies for design and analysis of small clinical trials, which are present in the evaluation of new therapies in rare diseases. We have shown how the IDeAl project has been providing answers for many of the questions, however several specific questions have to be solved over time. The EMA’s interests like extrapolation, standards of evidence, data‐driven decision‐making, understanding value of research, multidisciplinary, simulations, effects, randomisation, bias and the use of historical data are addressed by IDeAl’s research. These aspects are treated within the 10 workpackages of the IDeAl project, which are depict in figure 146.
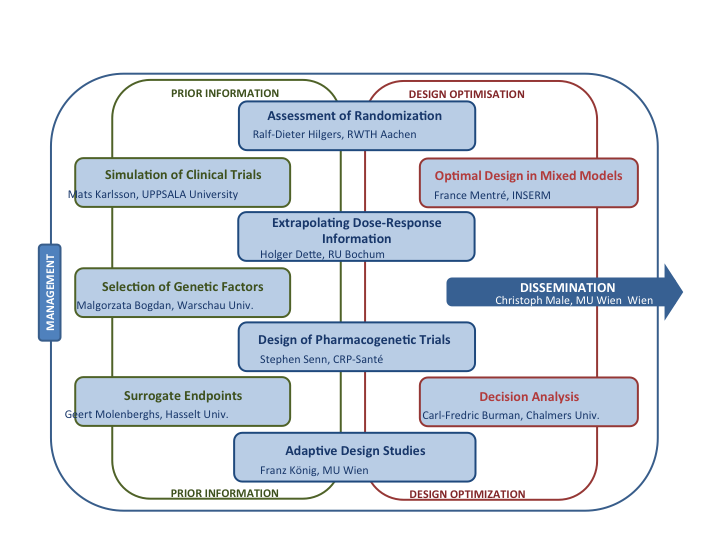
Figure 1: Exhibit of the IDeAl project broken down in the workpackages.
Overall, there are three levels which have to be addressed when considering small clinical trials.
At the first level, the rigorous application of already developed efficient design and analysis techniques is recommended. Using these methods can lead to application of techniques used in traditional clinical trials in smaller populations also. The benefit would be, that these techniques are in accordance with the regulatory and scientific guidelines and thus already accepted by all stakeholders. Within this context one can think about using optimal crossover designs, using ANCOVA models and avoiding analysis of percent change analysis, etc. At this level training about best methods and consultation forum for researchers and patients are most effective and necessary.
At the second level, evaluations of the traditional methods for design and analysis of clinical trials are necessary, to show the validity of these methods with respect to small sample sizes. Within this context, we need to understand e.g. when randomization fails to protect against bias, if linear mixed effects models are sensitive against imbalance, how reliable are interim data in an anyhow small clinical trial. With this in mind, all main stakeholders have to be well informed about, how “small” is “too small” and traditional methods fail.
And consequently at the third level, new methods should be developed for design and analysis of clinical trials where the traditional methods fail. The research of the IDeAl project is addressed in particular to level two and three. It is important not only to publish the research findings of the project in scientific journals. A strong mandate is to inform all relevant stakeholders through workshops, webinars etc about these methods and to train young scientist with these methods.
Randomization is one of the key features of clinical trials in drug development to minimize bias in clinical trials and consequently identify differences in the outcome variable by treatments alone. The argument is well accepted for larger trials but the less is known for smaller trials. Obviously, the question arises, which randomization procedure performs best for smaller clinical trials, and according to the ICH E6 guideline, what is the appropriate analysis method. In rare diseases there are two types of bias which might affect the outcome, selection and chronological bias. Meanwhile our analysis indicates that they are working in an opposite direction. Depending on the amount of bias there is no unique choice of the best procedure, however an analysis of the performance could be made using the software tool randomizR. After conducting a trial according to a specified randomization procedure, the appropriate statistical test is a randomization test. The implementation of this test in the software is currently under work.
Biostatisticians have frequently and uncritically accepted the measurements provided by their medical colleagues engaged in clinical research. Such measures often involve considerable loss of information. Particularly unfortunate is the widespread use of the so‐called 'responder analysis,’ which may involve not only a loss of information through dichotomization, but also extravagant and unjustified causal inferences regarding individual treatment effects at the patient level, and, increasingly, the use of the so‐called number needed to treat scale of measurement. Other problems involve inefficient use of baseline measurements, the use of covariates measured after the start of treatment, the interpretation of titrations and composite response measures. Many of these bad practices are becoming enshrined in the regulatory guidance to the pharmaceutical industry. We consider the losses involved in inappropriate measures and suggest that statisticians should pay more attention to this aspect of their work47.
It is well know that there is a considerable loss of information when continuous variables are dichotomised. In trials in common diseases, sample sizes are often greater than is necessary to provide proof of efficacy because trials are sized to prove safety and tolerability. Where this is the case, dichotomies, although still to be regretted, may not have a disastrous effect on the ability to prove efficacy. For rare diseases this will not be the case and such measures can and should be avoided48-51.
A regrettably common use of baseline measures is to construct so called change scores, or worse, calculate percentage change from baseline. The first does not make an efficient use of baselines and the second compounds this error by constructing a measure that has very poor distributional properties. There is scope for considerable gains in efficiency by using instead analysis of covariance (ANCOVA) fitting the baseline values or, where relative change is considered important, log transforming the baselines and outcomes prior to using ANCOVA49,52,53.
Especially when trials are small, considerable information can be gained by collecting measurements repeatedly over time. Moreover, such longitudinal profile allow the assessments of effect, largely based on within‐patient changes, that otherwise could not be studied. Partial longitudinal profiles offer well‐known opportunities when patients drop out from therapy or from the study altogether, prior to the planned end of the study54.
Stratification may or may not improve the efficiency of a trial by reducing the variance of the treatment effect. This is rather questionable, where the sample size is small and high unbalanced strata are to be expected. On the other hand, the argument for stratification is to reduce variance. This does not hold in general for rare diseases.
Adaptive interim analyses29 are another tool to improve the performance of clinical trials. However, the operating characteristics of potential adaptations should be carefully evaluated by clinical trial simulations beforehand. Especially adaptive seamless designs have a potential in small populations as they allow to tackle different objectives within a single trials using all (limited) data at hand.
RDH declares to have no relevant affiliation with any organisation or entity with a financial interest, direct or indirect, in the subject matter or materials discussed in the manuscript.
FK declares to have no relevant affiliation with any organisation or entity with a financial interest, direct or indirect, in the subject matter or materials discussed in the manuscript.
GM declares to have no relevant affiliation with any organisation or entity with a financial interest, direct or indirect, in the subject matter or materials discussed in the manuscript.
SS Acts as a consultant to the pharmaceutical industry and holds shares in Novartis. He maintains a full declaration of interest here http://www.senns.demon.co.uk/Declaration_Interest.htm. He is not aware however that any matters discussed here will have any material effect on any organisation or entity with whom he is associated.
Carl Fredrik Burmann (PhD, Chalmers University of Technology, Göteborg, Sweden )
Malgorzata Bogdan (PhD, Warschau University, Warschau, Polen)
Holger Dette (PhD, Ruhr University Bochum, Germany)
Ralf‐Dieter Hilgers (PhD, RWTH Aachen University, Germany)
Mats Karlsson (PhD, UPPSALA University, Uppsala, Sweden)
Franz König (PhD, Medical University Vienna, Austria)
Christoph Male (PhD, Medical University Vienna, Austria)
France Mentré (PhD, INSERM Paris, France)
Geert Molenberghs (PhD, I‐BioStat, KU Leuven, Leuven Belgium)
Stephen Senn (PhD, LIH Luxembourg, Luxembourg)
This research receives funding by grant 602552 from the European Union's 7th Framework Programme for research, technological development and demonstration under the IDEAL Grant Agreement no 602552.