
Effect of combined NPHS2 and ACTN4 variants in the onset and severity of focal segmental glomerulosclerosis
Michelle T. Passos1, Patricia Varela2, Danilo E. Fernandes1, M. Goretti Polito1, João B. Pesquero2#, Gianna Mastroianni Kirsztajn1#*
1Division of Nephrology, Department of Medicine, Federal University of São Paulo, São Paulo, Brazil
2Center for Research and Molecular Diagnostic of Genetic Diseases, Department of Biophysics, Federal University of São Paulo, São Paulo, Brazil
#Both authors had equal participation in this study and share senior authorship
Abstract
Introduction: Focal segmental glomerulosclerosis (FSGS) can be caused by mutations in the genes NPHS2, ACTN4, TRPC6, and INF2 among others, presenting variable levels of proteinuria, including nephrotic syndrome, that frequently progress to end-stage renal disease (ESRD). The establishment of the genotype-phenotype correlation caused by mutations in genes expressed in the podocyte could contribute to understanding their role in FSGS and to the decision-making in the clinical setting in similar cases.
Methods: Genomic DNA was extracted from peripheral blood of the proband, his brother, sister and mother. All the exons of the genes NPHS2, ACTN4, TRPC6, and INF2 were amplified by a polymerase chain reaction, purified, and sequenced by the Sanger method. The presence of variants was evaluated in the proband with FSGS and relatives, reviewed, and annotated using dbSNP and HGMD.
Results: In the clinical evaluation, the proband and his brother presented childhood-onset nephrotic syndrome, added with renal biopsies confirming FSGS, which was resistant to steroid and other immunosuppressive drugs, and progressed to ESRD. Both patients showed the variants p.P316S in NPHS2 and p.G894S in ACTN4, as well as the polymorphism p.R229Q in NPHS2, all variants in heterozygosis. Their parents were healthy, and the mother presented only the variant p.P316S in NPHS2 in heterozygosis.
Conclusions: The family members with FSGS had a combination of the variants p.P316S and p.R229Q in NPHS2, and p.G894S in ACTN4, shared similar clinical presentation, nephrotic syndrome with onset in late childhood that rapidly progressed to ESRD.
Introduction
Focal segmental glomerulosclerosis (FSGS) is a common cause of end-stage renal disease among children and adults with glomerulopathies. Most cases are primary or of unknown etiology; a few are secondary and less frequently genetic. The diagnostic resources available in daily practice do not distinguish all forms of this disease. Although genetic mutations are one of the known causes of FSGS, genetic testing is not easily available, and situations in which their use would be precisely indicated are not well established.
Several mutations in more than 50 genes have been implicated in the development of FSGS, including NPHS2, ACTN4, TRPC6, and INF2, among others, and such mutations are also present in some types of steroid-resistant nephrotic syndrome (SRNS) in childhood. In genetic FSGS as well as in SRNS, these mutations determine alterations in proteins (podocin, alfa-actinin 4, transient receptor protein channel 6, and inverted formin 2, respectively) that are essential to maintain the glomerular podocyte function among others.
Genetic forms of FSGS can occur in familial1-3 as well as in sporadic forms of this disease4. It is important to know the pathogenic mutations involved in each case, characteristics of disease presentation and course, as well as response to treatment, to gain information on the genotype-phenotype correlation and to contribute to a better disease management.
Materials and Methods
Ethical Approval and Informed consent
All procedures were approved by the research ethics committee of the Federal University of São Paulo and with the 1964 Helsinki declaration. Informed consent was obtained from all individual participants included in the study.
Patients
Patients with familial SRNS and their relatives were included. Those that would not provide informed consent would be excluded.
Thus, members of a family in which two boys had the histopathological diagnosis of FSGS were evaluated. In addition to renal function tests, a screening mutation analysis for NPHS2, ACTN4, TRPC6, and INF2 was performed in the proband 13 years old), his brother (22 years old), sister (21 years old), and mother (44 years old).
DNA Sequencing
Sanger sequencing was conducted as described by Riguetti et al. (2020)3. Briefly, genomic DNA was extracted from peripheral blood samples; the exons regions of the genes NPHS2, ACTN4, TRPC6 and INF2 were amplified by polymerase chain reaction (PCR) and each amplicon was purified and sequenced. Sequences were compared with the reference DNA, confirmed by reverse strand sequencing, and analyzed. Variants were reviewed and annotated using dbSNP (single-nucleotide polymorphism database (ncbi.nlm.nih.gov/projects/SNP/) and HGMD (Human Genome Mutation Database professional, biobaseinternational.com/product/hgmd).
Renal function markers
Creatinine (in urine and serum) was determined by alkaline picrate method, proteinuria (random and 24-hour proteinuria) by immunoenzymatic assay method, and albuminuria by immunoturbidimetry. Albumin/creatinine (ACR) and protein/creatinine ratios were determined and glomerular filtration rate based on serum creatinine levels was estimated by the Chronic Kidney Disease Epidemiology Study (CKD-EPI) equation.
Results
Familial FSGS report
The proband was a Caucasian boy that was asymptomatic until 13 years of age when proteinuria was detected in a regular check-up motivated by his family history of CKD (report of his older brother with NS due to FSGS and some relatives with ESRD of unknown etiology). His mother reported that he previously presented only frequent episodes of rhinitis. After this check-up he was evaluated by a nephrologist, presenting already nephrotic syndrome, with anasarca and even ascites.
At admission (13 years old), the proband had 24-hour proteinuria of 4.7g/24h and serum creatinine of 0.5 mg/dL. He was submitted to several laboratory tests to investigate eventually associated diseases that revealed: non-reagent anti-HCV, anti-HIV, HBsAg, ANA, anti-ds-DNA, ANCA, and normal levels of C3, C4, and CH50. The ultrasound showed kidneys with normal size and normal cortex-medulla differentiation, but with a mild increase in their echogenicity. He was then submitted to a kidney biopsy that diagnosed FSGS, mild tubulointerstitial lesion, and, by immunofluorescence, glomerular segmental deposits of IgM and C3.
The patient was initially treated with steroids. After four months of use of 1mg/kg/day of oral prednisone, he still presented nephrotic syndrome (24-hour proteinuria of 7.8g, serum albumin of 2.5 g/dL but his creatinine clearance was normal. Urinalysis showed 10 leukocytes/hpf and 25 erythrocytes/hpf, and the presence of erythrocyte dysmorphism.
He has also received diuretics, an inhibitor of angiotensin-converting enzyme, as long as possible, and statins. After steroid withdrawal, he was treated with cyclosporine without any significant decrease in proteinuria, and with mycophenolate sodium, without success.
Twelve months after diagnosis, his serum creatinine was 1.53 mg/dL and, twelve more months later, 2.40 mg/dL, while the levels of proteinuria varied between 6.0 and 12.0 g/24h and serum albumin, between 2.2 and 2.5 g/dL. He had hyperlipidemia and hyperuricemia. Along the third year of follow-up, sinusitis became frequent. By this time, the immunosuppressive treatment was interrupted. Afterward, he received general treatment for CKD for seven months until renal replacement therapy was necessary.
With regard to his family, the parents were not consanguineous. His brother had the diagnosis of FSGS at the age of seven years and developed ESRD after six years, being submitted to renal transplantation. His sister, mother and father were healthy. All of them were submitted to a renal evaluation and had a normal glomerular filtration rate and no urinary abnormalities.
Mutation analysis
DNA sequence analysis revealed the presence of three heterozygous variants in the patient and his brother, two in the NPHS2 (p.P316S and p.R229Q), and one in ACTN4 (p.G894S). Their mother had only the variant p.P316S in NPHS2 in heterozygosis and their sister had no alterations in these genes (shown in Figure 1A and Table 1). The electropherogram analysis demonstrates the genetic variants found that correspond to changes in the genes NPHS2 and ACTN4 (shown in Figure 1B).
Table 1: Sequencing analysis of NPHS2 and ACTN4 in a family with FSG
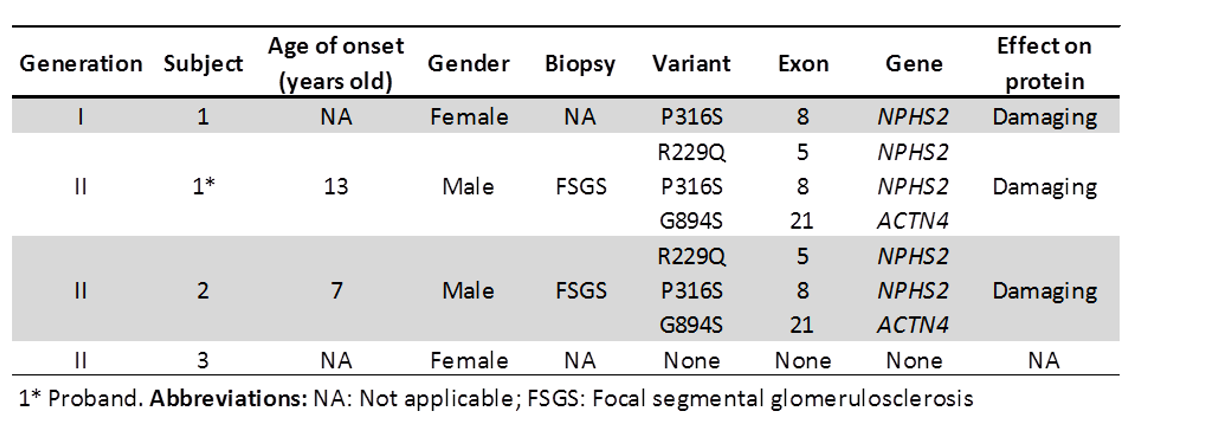
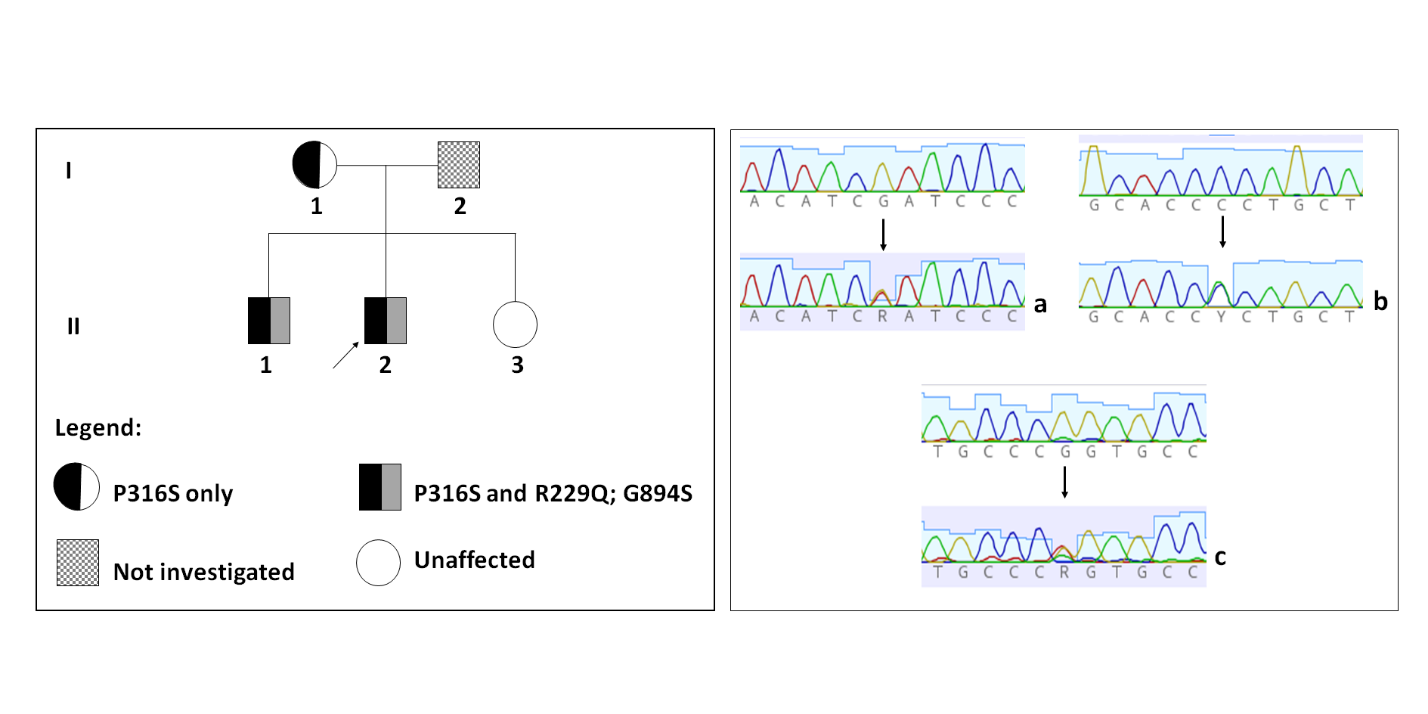
Figure 1: Genetic variants found in the family with FSGS.
Figure 1A: Pedigree showing affected members carrying the variants in the genes NPHS2 and ACTN4.
Figure 1B: Detection of variants in NPHS2 and ACTN4 by direct Sanger sequencing. Two heterozygous variants were detected in the NPHS2: the polymorphic variant p.R229Q (c.686G>A in exon 5) (a) and P316S (c.946C>T in exon 8) (b) and a new heterozygous variant detected in ACTN4, G894S (c.2680G>A in exon 21) (c).
Discussion
Here we evaluated two young brothers presenting with nephrotic syndrome secondary to FSGS. The proband presented the first symptoms at the age of 13 years. There was no response to steroids and subsequent immunosuppressive drugs used. Since FSGS was diagnosed, a gradual progression to ESRD occurred over three years. Both brothers showed the same genetic variants p.P316S (NPHS2) and p.G894S (ACTN4), as well as the polymorphism p.R229Q (NPHS2). Only the variant p.P316S was detected in their mother and no mutations in these genes were observed in their sister. Both mother and sister had a normal renal function.
The NPHS2 gene encodes the protein podocin. Mutations in this gene cause autosomal recessive SRNS as well as FSGS. Mikó et al. (2018) described the variant p.R229Q as the most commonly found NPHS2 mutation in the general population and as a human variant in which pathogenicity is dependent on another associated mutation5. We have previously demonstrated (Riguetti et al. 2020) that the association of the polymorphism p.R229Q (NPHS2) with p.R291W (NPHS2) led to late-onset familial FSGS3. Here we describe a different phenotype associated with both variants p.P316S and p.R229Q (NPHS2). The p.P316S (NPHS2) mutation was already described in homozygosity in patients with SRNS6,7.
It is of note that the p.R229Q variant is common in the European, South Asian, African and Latino-American population5; its population frequency is 0.02 (Brazilian Online Archive of Mutations - Abraom) and 0.03 (Gnomad/Exac), while the frequency of p.P136S was not found in any population frequency database.
In the present case, the affected family members carried an ACTN4 variant in addition to the combined NPHS2 mutations. The ACTN4 gene encodes the protein alpha-actinin 4. ACTN4 mutation causes most frequently autosomal dominant FSGS, which usually begins in adolescence or adulthood8, although also described in childhood in familial FSGS, as an isolated ACTN4 mutation9. It is characterized by non-nephrotic proteinuria and slow progression to ESRD8. Nonetheless, a case reported by Feng et al. (2016) describes an adolescent with collapsing glomerulosclerosis and rapid progression to ESRD8.
The G894S mutation (c.2680G>A, exon 21, ACTN4 gene) was first identified in the present study. However, its contribution to the severity of the phenotype of these patients is not clear, considering that the compound heterozygosity of the specific mutations spanning residues 270–351 and p.R229Q in NPHS2 by itself is sufficient to cause FSGS manifested as a nephrotic syndrome in children and adults3,10.
On the other hand, a bi-genic tri-allelic hit, a situation similar to our present report, was previously described in genetic FSGS, corresponding to a compound heterozygous mutations in NPHS2 and a heterozygous mutation in NPHS111. The cumulative effects of various mutations in different genes can cause FSGS or influence its severity12. For instance, according to Lennon et al. (2015), the coinheritance of COL4A5 mutations and homozygous MYO1E variants were associated with more severe kidney disease13.
It has been observed that mice carrying homozygous variants in the CD2AP gene develop FSGS with severe nephrotic syndrome and death within a few weeks after birth14. The individuals with variants in heterozygosis, on the other hand, have a slower progression, presenting only later some type of glomerular disorder15,16. In turn, Huber et al. (2006) demonstrated that bigenic heterozygosity in podocyte genes (CD2AP with Fyn or Synpo) in mice leads to the development of proteinuria and FSGS, while an isolated variant does not cause kidney disease15.
Löwik et al. (2008) identified one individual with FSGS mutations in heterozygosity in the NPHS2 and CD2AP genes, suggesting that the combined haploinsufficiency in two podocyte genes together might be responsible for the development of FSGS in humans11.
Concerning NPHS2 and ACTN4, no mutations have been described simultaneously in these two genes. However, it is difficult to ascribe the impact of the ACTN4 variant on the severity of the clinical picture presented by these family members, since the two variants p.R229Q and p.R291W in NPHS2 could be already sufficient to promote this phenotype.
Studies of families like this can contribute to a better understanding of the clinical presentation of different glomerulopathies caused by genetic mutations, thus establishing a correlation between their genotypes and phenotypes. In this family, the FSGS presentation in the affected members is not clinically different from that of primary FSGS in children and adolescents, except for the familial history of CKD and particularly of nephrotic syndrome. The affected brothers had no involvement of other systems in addition to the renal disease. In cases like this, it is important that the identification of genetic variants related etiologically to the development of FSGS is an available investigational tool for guiding the treatment of the patients and familial counseling.
In summary, we highlight here that combined gene defects in podocyte genes may play a role in the development of FSGS, as already shown by our group3 and others5,11. In addition, our findings suggest that the genotype described here is related to FSGS steroid-resistant and immunosuppressive drugs and could lead to ESRD in a short period.
Conflict of Interest
The authors have no conflicts of interest to declare.
Funding
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2014/27198-8 and 2019/05266-5) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, finance code 001).
Author Contributions
MTP, PV, DEF, and MGP collected data and performed the analyses under the supervision of JBP and GM-K. All the authors had access to the data and worked on the final version of this manuscript.
References
- Faubert PF, Porush JG. Familial focal segmental glomerulosclerosis: Nine cases in four families and review of the literature. American Journal of Kidney Diseases. 1997; 30(2): 265-70.
- Monteiro EJB, Pereira AC, Pereira AB, et al. NPHS2 mutations in adult patients with primary focal segmental glomerulosclerosis. J Nephrol. 2006; 19(3): 366-71.
- Riguetti MTP, Varela P, Fernandes DE, et al. Familial Focal Segmental Glomerulosclerosis With Late-Onset Presentation and R229Q/R291W Podocin Mutations. Front Genet. 2020; 11: 533373.
- Lipska BS, Iatropoulos P, Maranta R, et al. Genetic screening in adolescents with steroid-resistant nephrotic syndrome. Kidney International. 2013; 84(1): 206-13.
- Mikó Á, K. Menyhárd D, Kaposi A, et al. The mutation-dependent pathogenicity of NPHS2R229Q: A guide for clinical assessment. Human Mutation. 2018; 39(12): 1854-60.
- Ramanathan ASK, Vijayan M, Rajagopal S, et al. WT1 and NPHS2 gene mutation analysis and clinical management of steroid-resistant nephrotic syndrome. Mol Cell Biochem. 2017; 426(1-2): 177-81.
- Sadowski CE, Lovric S, Ashraf S, et al. A Single-Gene Cause in 29.5% of Cases of Steroid-Resistant Nephrotic Syndrome. JASN. 2015; 26(6): 1279-89.
- Feng D, Steinke JM, Krishnan R, et al. Functional Validation of an Alpha-Actinin-4 Mutation as a Potential Cause of an Aggressive Presentation of Adolescent Focal Segmental Glomerulosclerosis: Implications for Genetic Testing. PLoS ONE. 2016; 11(12): e0167467.
- Aucella F, De Bonis P, Gatta G, et al. Molecular Analysis of NPHS2 and ACTN4 Genes in a Series of 33 Italian Patients Affected by Adult-Onset Nonfamilial Focal Segmental Glomerulosclerosis. Nephron Clin Pract. 2004; 99(2): c31-6.
- Feltran LS, Varela P, Silva ED, et al. Targeted Next-Generation Sequencing in Brazilian Children With Nephrotic Syndrome Submitted to Renal Transplant: Transplantation. 2017; 101(12): 2905-12.
- Lowik M, Levtchenko E, Westra D, et al. Bigenic heterozygosity and the development of steroid-resistant focal segmental glomerulosclerosis. Nephrology Dialysis Transplantation. 2008; 23(10): 3146-51.
- Potter AS, Drake K, Brunskill EW, et al. A bigenic mouse model of FSGS reveals perturbed pathways in podocytes, mesangial cells and endothelial cells. PLoS ONE. 2019; 14(8): e0216261.
- Lennon R, Stuart HM, Bierzynska A, et al. Coinheritance of COL4A5 and MYO1E mutations accentuate the severity of kidney disease. Pediatr Nephrol. 2015; 30(9): 1459-65.
- Shih NY, Li J, Cotran R, et al. CD2AP localizes to the slit diaphragm and binds to nephrin via a novel C-terminal domain. Am J Pathol. 2001; 159(6): 2303-8.
- Huber TB, Kwoh C, Wu H, et al. Bigenic mouse models of focal segmental glomerulosclerosis involving pairwise interaction of CD2AP, Fyn, and synaptopodin. J Clin Invest. 2006; 116(5): 1337-45.
- Kim JM, Wu H, Green G, et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science. 2003; 300(5623): 1298-300.