
Fabry disease: Prototype of Lysosomal Storage Disorder with systemic involvement
Irene San-Román-Monserrat1*, Juan-Ramón Gimeno-Blanes2, María Elena Rodríguez-González-Herrero3, Andrea Sodi4, Alessandro Mecocci5, Marisol Alegría-Fernández6, David López –Cuenca7 and Beatriz Rodríguez-González- Herrero8
1Department of Internal Medicine, Hospital General Universitario Reina Sofía, Murcia, Spain
2Department of Cardiology, Hospital Universitario Virgen de la Arrixaca, Murcia, Spain
3Department of Ophthalmology, Hospital Universitario Virgen de la Arrixaca, Murcia, Spain
4Department of Ophthalmology, Careggi Teaching Hospital, Firenze, Italy
5Department of Informatics and Mathematical Sciences, Siena University, Siena, Italy
6Hospital Universitario Virgen de la Arrixaca, Murcia, Spain
7Hospital Universitario Virgen de la Arrixaca, Murcia, Spain
8ENT Consultant, Hospital Comarcal del Noroeste, Caravaca, Murcia, Spain
Fabry disease is a rare1 inherited metabolic disorder with systemic involvement. Heart, kidney, brain, skin and eye are the main involved systems, but virtually every tissue may be affected.
The disease is due to mutations in GLA gene, located in X chromosome (Xq22.1). GLA mutations result in a mutant α-Galactosidase-A enzyme (α-AGA) with deficient activity. α-AGA deficient activity leads to deposition of not-catabolised glycosphyngolipids, predominantly globotryaosyl ceramide (GB3) and, to a lesser extent, galabiosylceramide and group-B substances, originating systemic dysfunction2,3.
Contrary to other X-linked diseases, Fabry registries show that female carriers may develop clinical disease. Therefore, Fabry female patients may be not just asymptomatic carriers but also clinically suffer the disease4. Most widely accepted hypothesis to explain the existence of symptomatic female patients is that of random-inactivation of X chromosome in females or lyonization5. According to Lyon’s hypothesis in every cell from a female embryo one X chromosome is inactivated early in life. The key issue is that the inactivated X of a particular cell might be of either maternal or paternal origin, and this is randomly determined. If the inactivated X chromosome is the paternal one, the female embryo will probably express the pathological phenotype (inherited from her father in the daughter of a male carrier)6,7. Female patients from the family that we described in 2014, carrying p.M187R/g7219 T>G novel mutation, represent an exceptional example of how lyonization works8.
With our previous work from Medicina Clínica (2014) we reported on the largest Spanish Fabry family published to date with novel-mutation genetic diagnosis and comprehensive multidisciplinary evaluation (Figure 1). With this work we aim to update family clinical description, including two recently diagnosed members (male IV9, female IV10), to underline main clinical clues for early diagnosis and to recall the novel features we exposed in 2014.
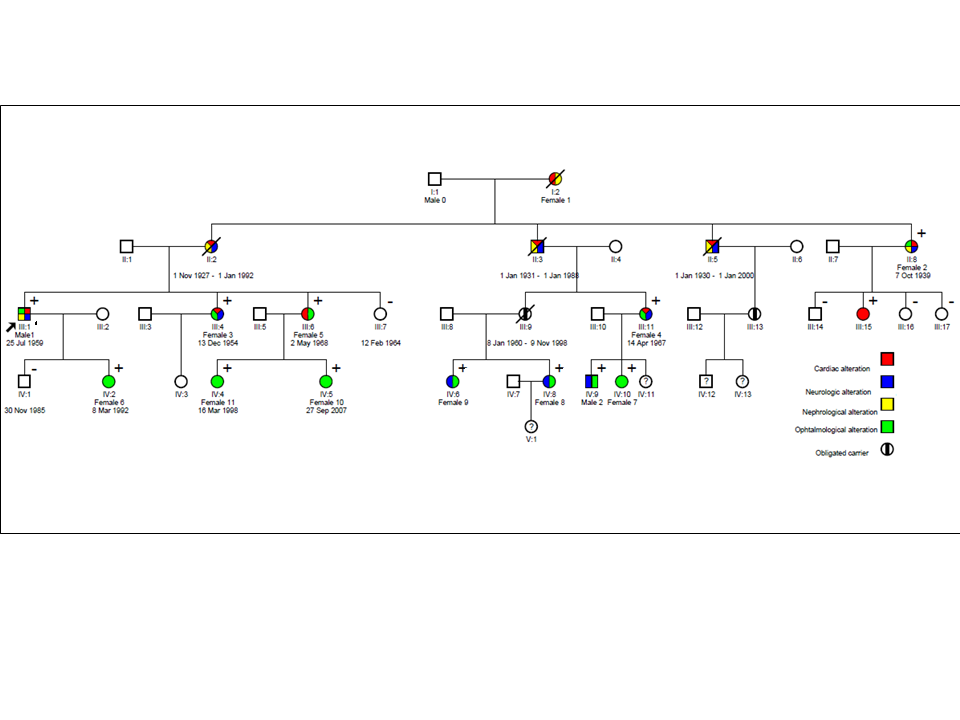
Figure 1: Family tree (updated 2016) Square means male, Circle means female, diagonal line means deceased + means carrier of p.M187R
Particularly novel was that systemic involvement and clinical severity were approached from a quantitative point of view, by means of Mainz Severity Score Index (MSSI) calculation in its classical and FOS versions(9). MSSI evaluates severity and systemic involvement using 4 subscores: general (maximum 18/76 or 13’5/64’5 points for classical and FOS versions respectively), cardiovascular (20/76 or 18/64’5 points), renal (18/76 or 18/64’5) and neurological (20/76 or 18/64’5 points). For every subscore severe involvement was considered if more than half of maximum points were obtained (>10/20 points for cardiac subscore, for example).
Another important feature exposed in our 2014 work was the description of novel mutation p.M187R/g7219T>G located in exon-4 of GLA gene. It is a missense mutation that affects methionine in position 187. Its absence in 100 healthy-population chromosomes was checked. Predictive in-silico analysis confirmed high probability of pathogenicity with elevated scores. Clinical co-segregation was concordant. There are 3 other reported missense mutations related to classic phenotype in this position (M187V, M187I, M187T)10-12, suggesting that this is a ‘‘hot spot’’ in GLA gene. In 2005 Matsuzawa et al built a structural mutated α-AGA13 model, in which 161 GLA gene mutations were analysed. Those mutations that involved three or more atoms, particularly if located in the main chain of α-AGA, tended to associate classical Fabry phenotype. This is the case of M187V and M187R mutations. This fact may at least partially explain in molecular terms why M187R mutation associates a severe phenotype.
Regarding systemic involvement 66% of individuals had 2 or more affected systems. 53% of individuals had cardiovascular disease, 53% had neurological involvement, 32% had renal involvement, 84% had other not-organ-specific involvement and 100% of individuals had ophthalmological manifestations (Table 1). These data clearly show the systemic nature of Fabry disease14.
| Males (n=4) | Females (n=15) | Males + Females | |
|---|---|---|---|
| Cardiovascular | 75% | 47% | 53% |
| Neurological | 100% | 40% | 53% |
| Renal | 75% | 20% | 32% |
| Ophthalmological* | 100%(n=2) | 100%(n=10) | 100%(n=12) |
| Other not organ specific | 100% | 80% | 84% |
Table 1. Systemic involvement in p.M187R carriers
*Only 2 males and 10 females were alive and willing to participate in our study about Fabry ophthalmopathy in the family
The heart was the main organ involved. 53% of individuals displayed Fabry cardiomyopathy, including young females. Cardiac involvement was early and severe and heart failure (HF) was the most frequent mode of death. 3 sudden deaths were reported. 2 females and 1 male died suddenly being 64, 63 and 57 years (yrs) respectively. Cardiac subscore of Fabry Outcome Survey-MSSI was severe for both males and females over 40yrs (16’3/18 in males vs. 13’6/18 in females). Cardiac involvement in female carriers was even more severe than in males in particular cases. We remark the case of female III4. Being only 59yr and 4yrs older than her brother (male III1 MSSIcardiac:12/20, diagnosis age 49yrs, α-AGA activity:9%, table3), she has even more severe cardiac findings. Both siblings had arterial hypertension (secondary to Fabry disease) and the proband was a light smoker in his youth. Female III4 displayed stage III HF secondary to severe NYHA III not-obstructive hypertrophic cardiomyopathy, with severe systolic impairment (Left Ventricle Ejection Fraction [LVEF]:30%-46%, severe ventricular asynchrony) and received cardiac resynchronization therapy plus implated cardiac defibrillator being 55yrs (MSSIcardiac:20/20, diagnosis age:53yrs, α-AGA activity:9% table3). This case exemplifies how lyonizaion may explain severe phenotype in Fabry females. It supports, together with the 3 above mentioned sudden deaths, the association of novel mutation with severe phenotype in both male and female carriers.
Our study was the first to report short PR-interval associated to delta wave, suggestive of accessory pathway. In this context, the occurrence of syncope deserves careful attention15: The combination of conduction disease, “Fabry-cardioneuropathy”, functional ischaemia and fibrosis secondary to advanced hypertrophic cardiomyopathy may trigger ventricular arrhythmia and sudden death16-18.
The storage of not catabolised ceramides in endocardial and myocardial cells partially explains the physiopathology of Fabry hypertrophic cardiomyopathy. Several recent works19 propose a more dynamic hypothesis for hypertrophy. Instead of simple passive endothelial storage of glycosphingolipids, a complex interaction of storage molecules (like Lyso-GB3), cytokines, inflammatory mediators implicated in fibrosis, apoptosis and in the proliferation of smooth-muscle fibers within vessel walls would happen. Aerts et al. found that, as opposed to GB3, left ventricular mass correlated with plasma Lyso-GB3 concentration in Fabry heterozygote females. They also obtained a trend to correlation between Lyso-GB3 increasing levels and higher MSSI punctuation, also only for heterozygote females20.
Cardiac-MRI was performed in those individuals with ECG or echocardiogram abnormalities/unclear findings (43%, 6/14 individuals that were alive at family diagnosis). Late gadolinium enhancement on cardiac-MRI was found in every tested individual, including a female with normal echocardiogram. This remarks the importance of cardiac-MRI for prognostic stratification of Fabry cardiomyopathy21-22. Cardiac disease was the most frequent mode of death in Fabry males and females23. Mortality directly attributable to Fabry disease in p.M187R carriers was 21% (4/19, 2males, 2females). In 100% of the deceased cases Fabry cardiomyopathy was present, with 3/4 sudden deaths. Finally, Fabry cardiomyopathy represents 1%24-25 of all unclear-origin genetic cardiomyopathies. Therefore, cardiologists dealing with hereditary cardiac disease should be aware of Fabry disease signs and symptoms.
Renal disease was the second cause of death in males after cardiac causes. 32% developed renal disease and 2 males required hemodyalisis at 48 and 45yrs. Bilateral parapyelic cysts were identified in proband, being an important diagnostic clue. Microalbuminuria with normal glomerular filtration rate (GFR) was detected in one female in the fourth decade, which together with subtle ECG abnormalities set up the indication for enzymatic replacement therapy (ERT) starting. Heart and kidney transplant were performed to one male being 57yrs due to stage-IV cardiac failure and end stage renal disease. Renal disease was less severe in females. Nevertheless, 20% of females had chronic kidney disease (CKD). Fabry disease is a poorly recognized cause of CKD and should be screened out in every proteinuric nephropathy of unknown origin, particularly when it associates parapyelic renal cysts26-31.
Cerebrovascular disease affected 53% of individuals. Peripheral neuropathy was common, with 100% of those with neurological disease displaying peripheral neuropathy. Ear-Nose-Throat area was involved in 16% of individuals. Stroke happened in 16% of individuals (2males, 1female). It represented the debut of overt organic disease in all of them, being 49, 45 and 61yrs respectively32. In male cases posterior cerebral artery territory was the main area of cerebrovascular disease. Fabry disease causes approximately 1’2% of strokes previously considered as cryptogenetic33. Therefore, Fabry disease should be considered in differential diagnosis of young-onset cerebrovascular ictal disease, particularly when it involves posterior territory and assocites systemic disease in renal or cardiovascular sites, as it happened in our proband.
Regarding psychological involvement 2/19 individuals (1%, females III4 and IV8) displayed chronic anxious symptoms exacerbated by Fabry disease. A recent review34 suggested a high prevalence of psychological disorders such as depression in Fabry patients. They obtained that 8%–17% of Fabry males scored within the mild-to-moderate ranges of Depression, Anxiety and Stress Scale as compared with 11% of male-controls. There were no differences observed between Fabry and control females. Cognitive impairment in Fabry patients is also a poorly addressed subject. In the above mentioned review Fabry males demonstrated slower speed of information processing, reduced performance on measures of executive functions and were more likely to show significant reductions. Conversely, Fabry females performed similarly to controls. Another review35 made correlational analyses suggesting a link between cognitive impairment and clinical measures of disease severity.
Ophthalmological involvement deserved particular attention in this family. Eye involvement was detected in every individual from this large family regardless gender. Corneal disease was particularly severe, with 100% of individuals displaying cornea verticillata. Slit-lamp exam cornea verticillata images were particularly striking, with bizarre and early forms (Figure 2). These bizarre images were detected not only in older than 40yrs individuals with organic overt disease but also in younger paucisymptomatic/asymptomatic individuals, regardless their gender. In fact, the youngest individual in whom cornea verticillata was detected was a 7yr old female. This remarks the utmost importance of ophthalmological examination for early diagnosis of Fabry disease. Furthermore, in our present research line we have explored the potential implications of ophthalmological exam in terms of prognostic stratification of Fabry patients. We have obtained interesting results that have been accepted for publication in Retina, the Journal of Retinal and Vitreous Diseases36-38.

Figure 2: Cornea verticillata. 26yr old asymptomatic p.M187R carrier cornea verticillata
Other organs and systems are frequently involved in Fabry disease, such as skin (angiokeratomata), gastrointestinal, pneumological or rheumatological system. We summarize these sites involvement in table 2. Once more, this fact remarks the systemic character of the disease39-47.
| Total | Male | Female | Comments | |
|---|---|---|---|---|
| Gastrointestinal | ||||
|
Colickyabdpain-diarrhoea Constipation-Hemorroids |
5/19 (26%) 3/19 (16%) |
2/4 (50%) 0/4 (0%) |
3/15 (20%) 3/15 (20%) |
Missdiagnosis of irritable bowels yndrome in F II2 |
| ENT area** | ||||
| Tinnitus | 4/19 (21%) | 1/4 (33%) | 3/15(20%) | Occasional/Episodic tinnitus |
| Vertigo | 3/19 (16%) | 1/4 (33’3%) | 2/15 (13%) | Occasional Rotatory vertigo |
| Hypoacusia/Audiometry | 5/19 (26%) | 3/4 (75%) | 2/15 (13%) |
M1Aud: RE: severe mixed hearing loss, LE: mod sensorineural hearing loss. Bilat osteoma aud ch .F2 Aud: Both ears hearing impairment at 4 KHz |
| Pneumological | ||||
| Bronchospasm | 4/19 (21%) | 3/4 (75%) | 1/15 (6%) | With lower respiratory tract infs mainly during second to fourth decade. |
| Rheumatological | ||||
| Arthralgia(large joints) | 7/19 (37%) | 3/4 (75%) | 4/15 (26%) | ankles, knees, wrists, shoulders |
|
Osteoporotic fractures Raynaud |
0/19 (0%) 0/19 (0%) |
0/4 (0%) 0/19 (0%) |
0/15 (0%) 0/19 (0%) |
No osteoporotic fractures/Raynaud identified until the date. |
| Endocrinological | ||||
|
2º HyperPTH CKD*confirmed 2º HyperPTH CKD*probable |
2/6 (33%) 2/6 (33%) |
1/4 (25%) 3/4 (75%) |
1/3 (33%) 2/3 (66%) |
No PTH available data of M II3, M II5, F I2 and F II2)(deceased). |
| Thyroid | 1/19 (5%) | 0/4 (0%) | 1/15 (6%) | Thyroid nodule excised to F III9 (third decade, no AP available). |
| DM-2 | 1/19 (5%) | 1/4 (25%) | 0/15 (0%) | On oral antidiabetics M II5 (deceased) |
| Fastingimpairedglycaemia | 2/19 (10%) | 1/4 25%) | 1/15 (6%) | On diet, F II8 |
| Neoplastic disease | ||||
| Breast carcinoma | 2/19 (10%) | 0/4 (0%) | 2/15 (13%) |
F II8: Ductal SLQ R breast ca (surgery+RTAug‘05). Remission. F III9:Breast ca+MTS deceased 38y |
| Infectious disease | ||||
| Lowerrespiratorytractinfs. | 4/19 (21%) | 3/4 (75%) | 1/15 (6%) |
With bronchospasm, mainlyduring third to fourth decade. Community adquired pneumoniae M2 (35y) |
| B-Hepatitis | 1/19 (5%) | 1/4 (25%) | 0/15 (0%) | M II3 (aged 51y |
| Acute Pielonephritis | 1/19 (5%) | 0/4 (0%) | 1/15 (6%) | F II2 (aged 61 y) |
| Recurrent tonsillitis | 1/19 (5%) | 0/4 (0%) | 1/15 (6%) | F II2 (infance) |
| Costal Zoster-Herpes (P) | 1/19 (5%) | 0/4 (33.3%) | 1/15 (6%) | F III4 (aged 48y) |
| Mumps | 1/19 (6.6%) | 1/4 (33.3%) | 0/15 (0%) | M II5 (aged 21y). |
Table 2: Other organs and systems involvement
| MaleIII1 | MaleIV9 | FemII8 | FemIII4 | FemIII6 | FemIII11 | FemIV6 | FemIV8 | |
|---|---|---|---|---|---|---|---|---|
| Age (yrs) | 55 | 21 | 75 | 59 | 46 | 47 | 36 | 33 |
| α-AGA % | 9 | 11 | 57 | 37 | 71 | 11 | 59 | 51 |
| MSSINov’14 | 50/76 | 11/76 | 55/76 | 41/76 | 13/76 | 14/76 | 9/76 | 4/76 |
Table 3. α-AGA activity and MSSI data from M187R carriers
Three family members are on ERT with biweekly iv infusion of agalsidase beta, 2 males (III1, proband, and IV9) and 1 female (III11) (Figure 1). Other 2 females with clear indication of ERT (III4 and III6) have been repeatedly offered but have refused until the date. All the individuals under ERT attend regularly the infusion sessions. When indicated, they are receiving secondary prevention therapy for renal, cardiovascular and/or cerebrovascular disease (angiotensin-converter-enzyme blockers, betablockers, antiagregants/anticoagulants and statins) as stated in current guidelines. ERT was started in proband soon after Fabry diagnosis in 2009, being 49yrs. He had just suffered right hemibulbar cerebral infarction, was diagnosed of stage III chronic kidney disease and NYHA III not-obstructive hypertrophic cardiomyopathy. As we described in our 2014 article, he suffers severe classic Fabry phenotype. After 7 years on ERT renal function seems to be stable with MDRD-4 GFR of 37ml/min as well as stable LVEF, on 52%. No other cerebrovascular events have occurred. Achroparestesia and arthralgia are controlled with symptomatic treatment and ophthalmological signs remain unchanged. Male IV9 (21yrs) was diagnosed with paucisymptomatic classical Fabry without overt organic disease and subtle ECG repolarization abnormalities. He started on ERT only 1 year ago, and no cardiac, renal or cerebrovascular events have been observed to date. His mother is female III11 (49yrs). Microalbuminuria with normal GFR was detected together with subtle electrocardiographic abnormalities setting up the indication for ERT starting, also approximately 6 months ago. GFR remains under normal range, no changes in microalbuminuria have been observed and no cardiovascular or cerebrovascular events have happened48.
To conclude, with our 2014 work we intended to remark Fabry disease systemic nature with a real clinical family-case. The main “red-flags” that should trigger Fabry disease diagnostic work-up were exemplified. We also remarked the high severity that novel p.M187R mutation seems to associate. A quantitative approach to systemic severity and systemic involvement was intended by means of MSSI calculation. Severity was particularly high for cardiac features and corneal disease was evident for both male and female carriers. Finally with this family we described the first 3cases in literature in which short-PR interval associated delta wave in electrocardiogram in Faby disease, suggesting the existence of accessory intracardiac conduction pathways.
Abbreviations
AP: anatomopathological; Aud: Audiometry; aud ch: auditory channels; bilat: bilateral; Ca: carcinoma DM-2: type 2 diabetes mellitus; HyperPTH CKD*: secondary to Chronic Kidney Disease Hyperparathyroidism* out of 4 cases of CKD; infs: infections; 28 KHz: kilohertz; LE: left ear; mod: moderate; MTS: metastases; P: probable; RE: right ear; RT: radiotherapy; SLQ: superolateral quadrant; yrs: years. **: ENT symptoms evaluable only in 12/15 individuals; Audiometry feasible only in Male III1 and female II8.
References
- Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, et al. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. 2006 Jul; 79(1):31-40.
- Shabbeer J, Yasuda M, Benson SD, Desnick RJ. Fabry disease: identification of 50 novel alpha-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations. Hum Genomics. 2006 Mar; 2(5):297-309.
- Germain DP, Shabbeer J, Cotigny S, Desnick RJ. Fabry disease: twenty novel alpha-galactosidase A mutations and genotype-phenotype correlations in classical and variant phenotypes. Mol Med. 2002 Jun; 8(6):306-12.
- Dobyns WB, et al. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. American Journal of Medical Genetics. 2004; 129A:136–143
- Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 1961 Apr 22; 190:372-3.
- Dobrovolny R, Dvorakova L, Ledvinova J, Magage S, Bultas J, Lubanda JC, et al. Relationship between X-inactivation and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the alpha-galactosidase A gene in the Czech and Slovak population. J Mol Med (Berl). 2005 Aug; 83(8):647-54.
- Elstein D, Schachamorov E, Beeri R, Altarescu G. X-inactivation in Fabry disease. Gene. 2012 Sep 1; 505(2):266-8.
- San-Román-Monserrat I et al. Comprehensive clinical evaluation of a large Spanish family with Anderson-Fabry disease, novel GLA mutation and severe cardiac phenotype. Med Clin (Barc). 2014; 142(11):497–504
- Whybra C, Kampmann C, Krummenauer F, Ries M, Miengel E, Miebach E, et al. The Mainz Severity Score Index: a new instrument for quantifying the Anderson- Fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin Genet. 2004; 65:299–307
- Altarescu GM, Goldfarb LG, Park KY, Kaneski C, Jeffries N, Litvak S, et al. . Identification of fifteen novel mutations and genotype-phenotype relationship in Fabry disease. Clin Genet. 2001; 60(1):46-51
- Sadick N, Thomas L. Cardiovascular Manifestations in Fabry Disease: A Clinical and Ebhocardiographic Study. Heart Lung Circ. 2007; 16(3):200-6.
- Shabbeer J, Yasuda M, Benson SD, Desnick RJ. Fabry disease: identification of 50 novel alpha-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations. Hum Genomics. 2006 Mar; 2(5):297-309
- Matsuzawa F, Aikawa S, Doi H, Okumiya T, Sakuraba H. Fabry disease: correlation between structural changes in alpha-galactosidase, and clinical and biochemical phenotypes. Hum Genet. 2005; 117(4):317-28.
- Marchesoni CL, Roa N, Pardal AM, Neumann P, Caceres G, Martinez P, et al. Misdiagnosis in Fabry disease. J Pediatr 2010 May; 156(5):828-31.
- Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart. 2007; 93:528–35.
- Eckart RE, Kinney KG, Belnap CM, Le TD. Ventricular fibrillation refractory to automatic internal cardiac defibrillator in Fabry’s disease. Review of cardiovascular manifestations. Cardiology. 2000; 94(3):208-12
- Igawa O, Miake J, Hisatome I. Ventricular tachycardias and dilated cardiomyopathy caused by Fabry disease. Pacing Clin Electrophysiol 2005 Oct; 28(10):1142-3.
- Ikari Y, Kuwako K, Yamaguchi T. Fabry’s disease with complete atrioventricular block: histological evidence of involvement of the conduction system. Br Heart J. 1992 Sep; 68 (3):323-5.
- Aerts JM, Groener JE, Kuiper S, Donker-Koopman WE, Strijland A, Ottenhoff R, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A. 2008 Feb 26; 105(8):2812-7.
- Rombach SM, Dekker N, Bouwman MG, Linthorst GE, Zwinderman AH, Wijburg FA, et al. Plasma globotriaosylsphingosine: diagnostic value and relation to clinical manifestations of Fabry disease. Biochim Biophys Acta 2010 Sep; 1802(9):741-8.
- Moon JC, Sachdev B, Elkington AG, McKenna WJ, Mehta A, Pennell DJ, et al. Gadolinium enhanced cardiovascular magnetic resonance in Anderson-Fabry disease. Evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J. 2003 Dec; 24(23):2151-5.
- Moon JC, Sheppard M, Reed E, Lee P, Elliott PM, Pennell DJ. The histological basis of late gadolinium enhancement cardiovascular magnetic resonance in a patient with Anderson-Fabry disease. J Cardiovasc Magn Reson. 2006; 8(3):479-82.
- Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry.
- Elliott P, Baker R, Pasquale F, Quarta G, Ebrahim H, Mehta AB, et al. ACES study group. Heart. 2011 Dec; 97(23):1957-60.
- Monserrat L, Gimeno-Blanes JR, Marin F, Hermida-Prieto M, Garcia-Honrubia A, Perez I, et al. Prevalence of fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2007 Dec 18; 50(25):2399- 403.
- Herrero-Calvo JA. Fabry's disease: a form of chronic renal disease that may be diagnosed and treated. Nefrología. 2008; 28(1):13-9.
- Ries M, Bettis KE, Choyke P, Kopp JB, Austin HA, III, Brady RO, et al. Parapelvic kidney cysts: a distinguishing feature with high prevalence in Fabry disease. Kidney Int. 2004 Sep; 66(3):978-82.
- Anderson-Fabry Disease in Chronic Kidney Disease Patients Not on Renal Replacement Therapy. clinicaltrials gov 2015 September 1 [cited 15 A.D. Oct 9];Available from: URL: https://clinicaltrials.gov/show/NCT00728364
- Thadhani R, Wolf M, West ML, Tonelli M, Ruthazer R, Pastores GM, et al. Patients with Fabry disease on dialysis in the United States. Kidney Int. 2002 Jan;61(1):249-55.
- Merta M, Reiterova J, Ledvinova J, Poupetova H, Dobrovolny R, Rysava R, et al. A nationwide blood spot screening study for Fabry disease in the Czech Republic haemodialysis patient population. Nephrol Dial Transplant. 2007 Jan;22(1):179- 86.
- Kleinert J, Kotanko P, Spada M, Pagliardini S, Paschke E, Paul K, et al. Anderson- Fabry disease: a case-finding study among male kidney transplant recipients in Austria. Transpl Int. 2009 Mar; 22(3):287-92.
- Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke. 2009 Mar; 40(3):788-94.
- Rolfs A BTZMea. High prevalence of Fabry disease in young stroke patients. Lancet. 2005; 366:1754-6.
- Bolsover FE, Murphy E, Cipolotti L, Werring DJ, Lachmann RH. Cognitive dysfunction and depression in Fabry disease: a systematic review.
- Sigmundsdottir L, Tchan MC, Knopman AA, Menzies GC, Batchelor J, Sillence DO. Cognitive and psychological functioning in Fabry disease.
- San-Roman I, et al. Retinal Fabry: Towards new prognostic factors Retina 00:1–12, 2016.(ahead pub)
- Sodi A, Ioannidis AS, Mehta A, Davey C, Beck M, Pitz S. Ocular manifestations of Fabry’s disease: data from the Fabry Outcome Survey. Br J Ophthalmol. 2007 Feb; 91(2):210-4.
- Sodi A, Guarducci M, Vauthier L, Ioannidis AS, Pitz S, Abbruzzese G, et al. Computer assisted evaluation of retinal vessels tortuosity in Fabry disease. Acta Ophthalmol. 2013 Mar; 91(2):e113-e119.
- Orteu CH, Jansen T, Lidove O, Jaussaud R, Hughes DA, Pintos-Morell G, et al. Fabry disease and the skin: data from FOS, the Fabry outcome survey. Br J Dermatol. 2007 Aug; 157(2):331-7.
- Palla A, Hegeman S, Widmer U, Straumann D. Vestibular and auditory deficits in Fabry disease and their response to enzyme replacement therapy. J Neurol. 2007; 254:1433–42.
- Wasik JS, Simon RW, Meier T, Steinmann B, mann-Vesti BR. Nailfold capillaroscopy: Specific features in Fabry disease. Clin Hemorheol Microcirc. 2009; 42(2):99-106.
- Brown LK, Miller A, Bhuptani A, Sloane MF, Zimmerman MI, Schilero G, et al. Pulmonary involvement in Fabry disease. Am J Respir Crit Care Med. 1997 Mar; 155(3):1004-10.
- Magage S, Lubanda JC, Susa Z, Bultas J, Karetova D, Dobrovolny R, et al. Natural history of the respiratory involvement in Anderson-Fabry disease. J Inherit Metab Dis. 2007 Oct; 30(5):790-9.
- Eng CM, Fletcher J, Wilcox WR, Waldek S, Scott CR, Sillence DO, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007 Apr; 30(2):184-92.
- Mehta A, Ricci R, Widmer U, Dehout F, Garcia de LA, Kampmann C, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004 Mar; 34(3):236-42.
- Horiuchi H, Saito N, Kobayashi S, Ota H, Taketomi T, Takaoka K. Avascular necrosis of the femoral head in a patient with Fabry’s disease: identification of ceramide trihexoside in the bone by delayed-extraction matrix-assisted laser desorption ionization-time-of-flight mass spectrometry. Arthritis Rheum. 2002 Jul; 46(7):1922-5.
- Faggiano A, Pisani A, Milone F, Gaccione M, Filippella M, Santoro A, et al. Endocrine dysfunction in patients with Fabry disease. J Clin Endocrinol Metab. 2006 Nov; 91(11):4319-25.
- Biegstraaten M, Arngrimsson R, Barbey F, Boks L, Cecchi F, Deegan PB, et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry Working Group consensus document. Orphanet J Rare Dis. 2015; 10:36.