
Gayet -Wernicke encephalopathy in non alcoholic patients: A serious complication
Ammouri W*, Harmouche H, Alaoui M, Tazi ZM, Maamar MM and Adnaoui M
Internal Medicine Department, CHU Ibn Sina, University of Medicine Rabat, Morocco
Abstract
Gayet-Wernicke encephalopathy is an acute, reversible neuropsychiatric emergency due to thiamine defciency. Chronic alcoholism is recognized as the most common cause of Wernicke’s encephalopathy, but other causes, including fasting/starvation and malnutrition, have been documented within the scientific literature. These causes may not be readily recognized by healthcare professionals and may lead to Wernicke’s encephalopathy being overlooked as a diagnosis when a nonalcoholic patient presents with classic signs and symptoms of the disorder.
The diagnosis of the disease is clinical and magnetic resonance imaging confirms the diagnosis by the presence of hypersignals most frequently in the periaqueductal level of thalami and mammilary bodies. Urgent and adequate thiamine replacement is necessary to avoid death or progression to Korsakoff syndrome with largely irreversible brain damage. Wernicke Korsakoff syndrome refers to a condition where features of Wernicke encephalopathy are mixed with those of Korsakoff syndrome. Although thiamine is the cornerstone of treatment of Wernicke encephalopathy, there are no universally accepted guidelines with regard to its optimal dose, mode of administration, frequency of administration or duration of treatment. We present recommendations for the clinical management of Gayet-Wernicke encephalopathy based on literature review.
Thiamine and its relationship to the development of WE
Thiamine is a water soluble vitamin that is absorbed in the jejunum by 2 processes. When the thiamin level in the small intestines is low, an active transport portal is responsible for absorption. When the thiamine concentration is high, a passive mucosal process takes place. The daily requirement is about 5 mg and is usually provided by the diet. The body cannot produce thiamine and can only store up to 30 mg of it in tissues. The half-life of thiamine is 9-18 days. Thiamine is mostly concentrated in skeletal muscles but also found in the brain, liver, and kidneys. It’s excreted by kidney. Thiamine may also be destroyed by alkaline conditions (pH > 8) and by heat. Vitamin C and citric acid may be used as reducing agents to prevent the destruction of thiamine2,6.
Thiamine is an important co enzyme for 3 enzymes: α-ketoglutarate dehydrogenase complex, pyruvate dehydrogenase complex, and transketolase (enzyme metabolism and degradation of glucose). It is also essential in maintaining osmotic gradients across cell membranes.
Thiamine insufficiency and deficiency of these enzymes caused a focal lactic acidosis, an alteration of the blood brain barrier and the production of free radicals with cell death by necrosis and apoptosis1,4. This damage begins as early as 4 days after thiamine deficiency and steadily progresses in severity, eventually leading to programmed cell death. After 14 days of deficiency, lesions develop on the brain. These lesions and the neurological impairment that they cause are considered the classic signs and symptoms of WE. In the case of thiamine deficiency, the disorder is rapidly increased by the introduction of sweet food or infusion of intravenous glucose solution without vitamin supplementation (brutal consumption of thiamine body reserves via participation in glucose metabolism).
Anatomical lesions are located in the periventricular regions around the third and fourth ventricles, the aqueduct of Sylvius and the mammillary bodies. Also, Thiamine plays a key role in intracellular glucose metabolism and it is thought that thiamine inhibits the effect of glucose and insulin on arterial smooth muscle cell proliferation. Inhibition of endothelial cell proliferation may also promote atherosclerosis. Endothelial cells in culture have been found to have a decreased proliferative rate and delayed migration in response to hyperglycemic conditions. Thiamine has been shown to inhibit this effect of glucose on endothelial cells.
The pathology reflects an acute and massive vitamin B1 deficiency due to either its reduced consumption or absorption, or because of alcohol use4,6.
WE is considered a disease of adult alcoholics, but it can also be found in other scenarios, such as in the chronically undernourished and cachectic patients suffering from cancer or HIV. Also, recent cases are reported as a complication of hyperemesis gravidarum5and restrictive bariatric surgery. Thiamine deficiency in infants was also reported in the pediatric population and in non-alcoholic conditions. Predisposing conditions such as magnesium deficiency and defects in the SLC19A3 gene causing thiamine transporter-2 deficiency, malignancies, intensive care unit stays and surgical procedures for the treatment of obesity or a defective soy-based formula were described in children and adolescents with WE9-12.
Current issues regarding diagnosis and treatment of Gayet -Wernicke encephalopathy
The clinical diagnosis of WE is missed in 75–80% of cases1–3,13. The classical triad of gait ataxia, eye signs (nystagmus, ophthalmoplegia) and global confusion, as described by Karl Wernicke n 1881, is seen in only 16–20% of patients1,2,14. More rarely, a decrease in deep tendon reflexes, peripheral polyneuropathy and decreased tone or dysarthria were observed15. To compound the problem, Wernicke encephalopathy is not only dif?cult to differentiate from drunkenness and other causes of confusion, but it also often coexists with other disorders that cause confusion, such as alcohol withdrawal, benzodiazepine withdrawal, sepsis, hypoxia, hepatic encephalopathy and head injury. Due to the myriad of clinical features of WE, the diagnosis of the disorder may be difficult and other investigators have made valuable suggestions on how to address the challenge. Caine et al16, developed what are known as operational criteria for the diagnosis of WE. Based on these criteria, WE should be diagnosed when 2 of the following 4 signs are present: dietary deficiency, oculomotor abnormalities, unsteadiness of stance and gait, and either altered mental state or mild memory impairment. Also, Thomson et al17 have identified and classified other clinical signs and symptoms of thiamine deficiency based on 15 studies published over a 125-year period. For nonalcoholic malnourished patients, these signs and symptoms depend on the magnitude of the WE, whether it is mild, moderate, or severe
|
Mild Wernicke’s Encephalopathy |
Moderate Wernicke’s Encephalopathy |
Severe/Terminal Wernicke’s Encephalopathy |
|
Anorexia Nausea/vomitingVisual alterations |
Short-term memory loss Apathy/anxietyApprehension Emotional changes |
Disorientation ConfabulationOnset of coma |
Table 1: Categorization of frequent signs and symptoms of Wernicke’s Encephalopathy16
In the guidelines for the management of WE published by the European Federation of Neurological Societies, Galvin et al18 note that signs of vomiting and dietary deficiency are more common in nonalcoholics, and impaired eye movements and motor function are more common in alcoholics. In addition, alcoholics present with the classic triad of symptoms more frequently than non-alcoholics.
Also, Non-alcohol-related WE was more often associated with female gender, younger age, shorter duration of precipitating illness and better survival rate compared to alcohol-related WE19.
Laboratory and radiographic testing may be used to support and confirm the clinical diagnosis of WE, but they are not recognized as methods for diagnosing WE1,6.
To assess the concentration of available thiamine in the bloodstream, the blood, should be collected prior to thiamine administration (it is highly influenced by the recent thiamine intake) and kept away from light (thiamine is degraded in neutral and alkaline solutions, even at room temperature). We recommend surrounding the tube with aluminum foil and place it in a container cooled by ice until its delivery to the laboratory.
The presence of low total thiamine or thiamine diphosphate levels is neither necessary nor sufficient for diagnosis. Because, it does not accurately represent body thiamine status and contain only a fraction of total body thiamine3,6. The measurement of 24 hours urine thiamine levels has little utility in the diagnosis of WE. But due to practical difficulties, it’s not commonly used in clinical practice. It has low specificity for WE and the urine thiamine level provides information about the adequacy of thiamine intake, but does not quantify tissue thiamine store.
Other laboratory tests provide an indirect estimation of thiamine status, including the thiamine pyrophosphate effect. The thiamine pyrophosphate effect measures erythrocyte ketolase activity when thiamine diphosphate is added to serum in vitro. This is an actual representation of tissue thiamine level. Measurement of red blood cell transketolase activity provides a measure of thiamine status. However, this is not routinely available on emergency basis, and it is more important to make a presumptive diagnosis of WE and to treat the patient as soon as possible6.
As clinical diagnosis is sometimes difficult when the triad of WE is incomplete, imaging may be particularly important for confirming the syndrome. MRI remains the investigation of choice by showing T2 hyperintensities in pariventricular regions (Figure 1). MRI is still probably the best method for confirming the diagnosis, particularly using a FLAIR sequence. These lesions are typically found in the thalamus, mammillary bodies, around the third ventricle, tectal plate, and periaqueductal area, in alcoholics with WE and throughout other regions of the brain in nonalcoholics with WE. However, as such signal abnormalities are not pathognomonic (93% specificity)7,8,20. It therefore remains necessary to eliminate the differential diagnoses (such as stroke, cerebral venous thrombosis, acute encephalitis and cerebral lymphoma).
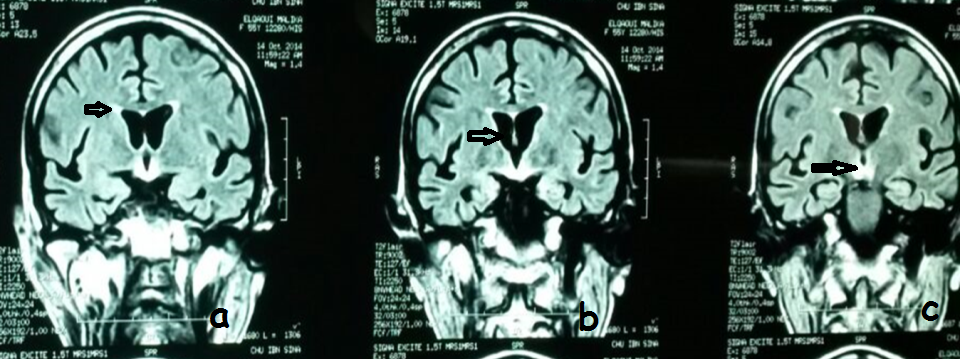
Figure 1: Coronal FLAIR-weighted brain magnetic resonance imaging (patient presented a Gayet Wernicke encephalopathy as a complication of gastrointestinal involvement of HIV disease shows bilateral symmetrical signal increases in the periventricular region, mammillary bodies, thalami (left) and periaqueductal area.
WE is considered a medical emergency. Early identification and immediate intervention, as outlined are crucial to producing positive patient outcomes. Even in the absence of a confirmed diagnosis, it is advised that clinicians initiate interventions in suspected cases. The treatment is based on parenteral thiamine therapy (vitamin B1). Different protocols have been proposed, for some to stop vomiting and resuming a normal diet15. The lack of randomized controlled trials precludes the establishment of strong evidence-based guidelines for thiamine administration. The current published guidelines are based primarily on case reports. While no consensus exists as to the ideal thiamine dose and form or duration of treatment, both the Royal College of Physicians3,6,18 and the European Federation of Neurological Societies18 have published guidelines for the treatment of WE in alcoholics. These guidelines must also serve as the basis for the management of WE in nonalcoholics since no published guidelines specific to this population exist. In patients who are identified as at risk for developing WE, who display WE-like symptoms, or who are diagnosed with WE, immediate high-dose thiamine administration either intravenously (IV) or intramuscularly (IM) is recommended to raise the blood concentration and cellular uptake in a timely manner2,6,14.
Time is of the essence in reversing and preventing neurologic damage. For at-risk patients, free thiamine of 250 mg (ampoules of 250mg per 2 ml) plus additional B-complex vitamins (250 mg thiamine, 4 mg riboflavin, 160 mg niacin, 50 mg vitamin B 6) and 500 mg of vitamin C should be infused IV daily for 3 to 5 days. An alternative approach is an IM injection of 250 mg thiamine daily for 3 to 5 days.
In patients with either a confirmed or a suspected diagnosis of WE, the recommendations using thiamine are consensual: high doses of parenteral thiamine. Dosages will vary slightly from reference to the other of 100 mg (ampoules of 100mg/ml) per day to more than 500 mg (two ampoules of 250mg/2ml) three times daily, the intravenous route being preferred for intramuscular administration21.
Free thiamine plus additional B-complex vitamins (50mg vitamin B6, 4 mg riboflavin, 150 mg niacin) and vitamin C (500 mg daily), should be infused IV 3 times daily for 3 days. Some patients may require up to 1 g of thiamine within the first 12 hours22. In addition, some clinicians recommend very initial high doses of parenteral thiamine (500–1500 mg daily) to enable diffusion of thiamine across the blood brain barrier to restore vitamin status21, prevent irreversible brain damage23, improve clinical signs6,22,24 and prevent death. Depending on the cause of WE, there may be other electrolyte abnormalities therefore need to be assessed and deranged levels corrected diligently. Serum electrolytes therefore need to be assessed and deranged levels corrected diligently. Also, magnesium is an essential cofactor in many thiamine dependent enzymes, and low levels of magnesium have been implicated in thiamine de?ciency and Wernicke encephalopathy and failure to respond to parenteral thiamine replacement. So, patients may require daily supplementation with magnesium (10–30 mEq), potassium (60–180 mEq), and phosphate (10–40 mmol). If improvement is observed after the 3 days, either IV infusion or IM injection should continue with 250 mg of thiamine plus B-complex and vitamin C once daily for 5 additional days or until no further improvement is experienced by the patient (Table2). At this point, the patient may be transitioned to oral administration of 100 mg thiamine 3 times daily2.
|
Treatment of patients with a de?nitive diagnosis of Wernicke encephalopathy Wernicke |
At least 100–500 mg intravenous thiamine three times daily for 3 days, and 250 mg once daily for 5 days.The treatment is then followed by oral thiamine, 100 mg 3 times daily for 1-2 weeks. |
|
Prophylactic treatment of patients with suspected or at risk of Wernicke encephalopathy |
At least 100–200 mg three times daily intramusculary or intravenous for 3–5 days, followed by oral thiamine,100 mg three times daily for 1–2 weeks and 100 mg daily thereafter |
Table 2: Recommended thiamine regimen for patients with Wernicke encephalopathy
No specified time frame for oral supplementation has been defined. The oral administration of thiamine is ineffective in correcting the moderate to severe thiamine deficiency seen in patients with WE. But, oral supplementation is recommended for the prevention and maintenance stages of WE. Otherwise healthy patients with mild thiamine deficiency should consume 30 mg6,20,22thiamine 3 times per day.
Prognosis
The prognosis of WE depend on the stage of the disease and prompt institution of thiamine. Evolution can include the full reversibility of disorders, motor sequelae, Korsakoff’s syndrome, coma and even death 1. The mortality rate ranges from 20 to 30%25.
Korsafoff’s syndrome can follow or accompany WE. It is a chronic largely irreversible sequela of untreated, or inadequately treated Wernicke encephalopathy, is characterised by dense anterograde amnesia and short-term memory loss associated with compensatory confabulation, with relative preservation of long-term memory and other cognitive skills.
Korsakoff syndrome, a chronic largely irreversible sequela of untreated or inadequately treated Wernicke encephalopathy, is characterised by dense anterograde amnesia and short-term memory loss associated with compensatory confabulation, with relative preservation of long-term memory and other cognitive skills.
Korsakoff syndrome is dif?cult to differentiate from other causes of dementia, and 20% of cases will require long term institutionalised care. Korsakoff syndrome preceded by, or occurring concurrently with acute Wernicke encephalopathy due to repeated episodes of clinical or subclinical thiamine de?ciency, results in Wernicke Korsakoff syndrome1,3.
Conclusion
WE is a medical emergency that requires intravenous thiamine administration as soon as the diagnosis is made to prevent any neurological sequelae, including Korsakoff syndrome. This rare complication in non-alcoholic patients deserves to be better known by clinicians. Preventive treatment of patients at risk of thiamine deficiency may prevent fatal consequences.
References
- Harper CG, Giles M, Finlay-Jones R. Clinical signs in Wernicke Korsakoff complex: a retrospective analysis of 131 cases diagnosed at autopsy. J Neurol Neurosurg Psychiatry. 1986 Apr; 49(4):341-5.
- Harper CG, Sheedy DL, Lara AI, Garrick TM, Hilton JM, Raisanen J. Prevalence of Wernicke-Korsakoff Syndrome in Australia: has thiamine forti?cation made a difference? Med J Aust. 1998 Jun 1; 168(11):542-5.
- Thomson AD, Cook CC, Touquet R, Henry JA. The Royal College of Physicians report on alcohol: guidelines for the managing Wernicke’s encephalopathy in the accident and emergency department. Alcohol Alcohol. 2002 Nov-Dec; 37(6):513-21.
- Torvik A. Wernicke encephalopathy: prevalence and clinical spectrum. Alcohol Alcohol. 1991; (Suppl 1): 381–4.
- Ngene NC1, Moodley J. Clinical awareness for health care professionals: Fatal encephalopathy complicating persistent vomiting in pregnancy. S Afr Med J. 2016 Jul 8; 106(8):792-4.
- Galvin R, Brathen G, Ivashynka A, Hillbom M, Tanasescu R, Leone MA. EFNS Guidelines for diagnosis, therapy and prevention of Wernicke encephalopathy. Eur J Neurol. 2010 Dec; 17(12):1408-18.
- Zuccoli G1, Santa Cruz D, Bertolini M, Rovira A, Gallucci M, Carollo C, et al. MR imaging findings in 56 patients with Wernicke encephalopathy: nonalcoholics may differ from alcoholics. AJNR Am J Neuroradiol. 2009 Jan; 30(1):171-6
- Ha ND1, Weon YC, Jang JC, Kang BS, Choi SH. Spectrum of MR imaging findings in Wernicke encephalopathy: are atypical areas of involvement only present in nonalcoholic patients?. AJNR Am J Neuroradiol. 2012 Aug; 33(7):1398-402.
- Isenberg-Grzeda E, Alici Y, Hatzoglou V, Nelson C, Breitbart W. Nonalcoholic Thiamine-Related Encephalopathy (Wernicke-Korsakoff Syndrome) Among In patients With Cancer: A Series of 18 Cases. Psychosomatics. 2016 Jan-Feb; 57(1):71-81
- Valevski AF, Kesler A, Sela BA, et al. Outbreak of life-threating thiamine deficiency in infants in Israel caused by a defective soy based formula. Pediatrics. 2005; 115(2):e233-8.
- Lallas M, Dejai J. Wernicke encephalopathy in children and adolescents. World J Pediatr . 2014; 10(4):293-8.
- Bohan PK, Yonge J, Connelly C, Watson JJ, Friedman E, Fielding G. Wernicke Encephalopathy after Restrictive Bariatric Surgery. Am Surg. 2016 Apr; 82(4):73-5
- Sechi GP, Serra A. Wernicke’s encephalopathy – new clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007; 6: 442–55
- Thomson AG, Marshall EJ. Wernicke’s encephalopathy: role of thiamine. Pract Gastroenterol. 2009; 33(6):21-22, 24-26, 28-30.
- Naidoo DP, Bramdev A, Cooper K. Wernicke's encephalopathy and alcohol-related disease. Postgrad Med J. 1991 Nov; 67(793):978-81.
- Caine D, Halliday GM, Kril JJ, Harper CG. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry. 1997; 62(1):51-60.
- Thomson AD, Cook CC, Guerrini I. Wernicke’s encephalopathy: ‘Plus ca change, plus c’est la meme chose.’ Alcohol Alcohol. 2008; 43(2):180-186.
- Sechi G, Sechi E, Fois C, Kumar N. Advances in clinical determinants and neurological manifestations of B vitamin deficiency in adults. Nutr Rev. 2016 May; 74(5):281-300.
- Scalzo SJ, Bowden SC, Ambrose ML, Whelan G, Cook MJ. Wernicke-Korsakoff syndrome not related to alcohol use: a systematic review J Neurol Neurosurg Psychiatry. 2015 Dec; 86(12):1362-8.
- Guler A, Alpaydin S, Sirin H, Calli C, Celebisoy N. A non-alcoholic Wernicke's encephalopathy case with atypical MRI findings: Clinic versus radiology. Neuroradiol J. 2015 Oct; 28(5):474-7.
- Boulanger AS, Paquette I, Létourneau G, Richard-Devantoy S. Thiamine et encéphalopathie de Gayet-Wernicke : quelles règles de prescription ? L'Encéphale, In Press, Corrected Proof, Available online 9 September 2016.
- Smithline HA, Donnino M, Greenblatt DJ. Pharmacokinetics of high-dose oral thiamine hydrochloride in healthy subjects. BMC Clin Pharmacol. 2012; 12:4.
- Cook CC, Hallwood PM, Thomson AD. Vitamin deficiency and neuropsychiatric syndromes in alcohol misuse. Alcohol Alcohol. 1998 Jul-Aug; 33(4):317-36.
- Tallaksen CM, Bell H, Bøhmer T. Kinetics of thiamin and thiamin phosphate esters in human blood, plasma and urine after 50 mg intravenously or orally. Alcohol Clin Exp Res. 1993 Jun; 17(3):712-6.
- Lana-Peixoto MA1, Dos Santos EC, Pittella JE. Coma and death in unrecognized Wernicke's encephalopathy: An autopsy study. Arq Neuropsiquiatr. 1992 Sep; 50(3):329-33.