
Genetic Landscape of aHUS: A Comprehensive Analysis of Genetic Variants Reported in The Literature
Rui-Ru Ji1*, Tatiana Serebriyskaya2,3, Natalia Kuzkina2,3
1Alexion Pharmaceuticals, Inc., 121 Seaport Boulevard, Boston, MA 02210, USA
2EPAM Systems, 22/2 Zastavskaya Street, MegaPark, 196084, Saint-Petersburg, Russia
3Moscow Institute of Physics and Technology, School of Biological and Medical Physics, 9 Institutskiy per., Dolgoprudny, 141701, Moscow, Russia
Abstract
Genetic information provides important guidance for long-term management of patients with atypical hemolytic uremic syndrome (aHUS), an extremely rare disease that primarily affects a patient’s kidney. To better understand the phenotypic impact of variants identified in aHUS patients, we systematically mined the National Library of Medicine database for case studies of aHUS patients with identifiable genetic variants. Allelic variants from 10 genes (C3, CFB, CFH, CFI, CFHR1, CFHR3, CFHR5, DGKE, CD46/MCP, and THBD) associated with aHUS were collected from 1652 patients. We analyze the enrichment of genetic variants in this “literature cohort” compared with a reference population, the Genome Aggregation Database (gnomAD). We also used a number of tools to predict the pathogenicity of the variants, attempting to reconcile all the results using the protein structure and conservation data. In total, we identified 447 unique genetic variants: 301 of these were not present in the gnomAD database and thus have “moderate” evidence of pathogenicity; 33 variants have “strong” evidence of pathogenicity by enrichment analysis. This study showcases an in silico framework that patient data aggregation and a large scale sequencing database provided a novel opportunity to understand genotype-phenotype associations in aHUS. This framework can be efficiently applied to other rare diseases where data are sparse to help improve the diagnosis and management of these patients.
Abbreviations
aHUS: atypical hemolytic uremic syndrome; gnomAD: Genome Aggregation Database; CD46/MCP: cluster of differentiation 46/membrane cofactor protein; CFH: complement factor H; CFI: complement factor I; CFB: complement factor B; C3: complement component 3; ACMG: American College of Medical Genetics; AF: allele frequency; CFHR1: complement factor H-related protein 1; CFHR3: complement factor H-related protein 3; CFHR5: complement factor H-related protein 5; DGKE: diacylglycerol kinase epsilon; THBD: thrombomodulin; MEDLINE: Medical Literature Analysis and Retrieval System Online; VEP: variant effect predictor; SIFT: sorts intolerant from tolerant substitutions; PROVEAN: protein variation effect analyzer; FATHMM: functional analysis through Hidden Markov Models
Introduction
Atypical hemolytic uremic syndrome (aHUS) is a disease characterized by the triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury.1 aHUS is extremely rare. Although the precise incidence and prevalence are unknown, it is estimated that aHUS affects 1–2 individuals per million inhabitants in the United States.2 aHUS can occur at any age, from the neonatal period to adulthood, although the onset appears to be more frequent in childhood than adulthood.3 In childhood, aHUS affects males and females equally.4 In adulthood, however, there is a preponderance in females affected and pregnancy can be a triggering event.5
aHUS is caused by chronic, uncontrolled activation or dysregulation of the alternative pathway of complement.6 Unlike the classical and lectin pathways of complement, the alternative pathway is continually activated and closely regulated by a number of complement regulatory proteins (such as complement factor H [CFH], complement factor I [CFI], and CD46/membrane cofactor protein [MCP]), so that host cells are not damaged.3 In most patients with aHUS, it has been demonstrated that the excessive activation of complement can result from mutations in any of the regulatory proteins or production of neutralizing autoantibody of these proteins, for example, anti-factor H antibodies.7,8
Although not required for diagnosis of aHUS, information on pathogenic complement mutations can be used to establish the cause of disease with accuracy, as well as to guide long-term disease management decisions and effective treatment.9-11 Approximately 20% of patients experience extrarenal manifestations, including central nervous system, cardiac, gastrointestinal, distal extremity, and severe systemic organ involvement.4,8,10 The ongoing risk of these manifestations varies among genotypes. For example, patients having mutations in C3, complement factor B (CFB), and CFH are particularly at risk for developing cardiovascular complications.12
In approximately 30–50% of individuals with aHUS, no mutation in a complement gene and no autoantibodies can be detected.7,8 However, recent literature exploring larger panels and new high-throughput sequencing technology show that other genes and pathways such as the coagulation pathway are also associated with aHUS.13 Nonetheless, many variants identified in patients are classified as variants of unknown clinical significance.14 Interpretation of these variants is difficult because: (1) some functional assays may be less reliable in predicting the impact of variants on protein function than others; (2) the rarity of the disease hinders the aggregation of a sufficiently large patient cohort; (3) incomplete penetrance is the norm in these patients.14,15
The standards and guidelines published in 2015 by the American College of Medical Genetics (ACMG) lay out an extensive framework of evidence for interpretation of sequence variants, including guidance for using population data and computational and predictive tools.14 By definition, causal genetic variants are enriched in patient populations as compared with reference populations. This principle has been exploited in genome-wide association studies to identify genes implicated in common diseases.16 In rare diseases, similar analyses can be utilized to assess pathogenicity of rare genetic variants if their allele frequencies (AFs) in patient populations and reference/control populations can be estimated. Large population databases such as the Genome Aggregation Database (gnomAD),17,18 containing 123 136 exome sequences and 15 496 whole-genome sequences of unrelated individuals from a wide variety of large-scale sequencing projects, now make it possible to obtain AFs of rare variants in reference populations. However, lack of a large patient cohort makes estimating variant AFs in patients with rare diseases challenging.
To overcome this difficulty, we created a “literature cohort” of more than 1600 patients with aHUS by compiling all case studies in MEDLINE. The allelic variants of 10 genes (namely, C3, CD46/MCP, CFB, CFH, complement factor H-related protein 1 [CFHR1], complement factor H-related protein 3 [CFHR3], complement factor H-related protein 5 [CFHR5], complement factor I [CFI], diacylglycerol kinase epsilon [DGKE], and thrombomodulin [THBD]) known to be associated with aHUS,3,19 along with the patient characteristics, were compiled and stored together in a relational database. We assessed the pathogenicity of genetic variants in the 10 genes with bioinformatic, statistical, and structural analyses.
Materials And Methods
Literature Mining and Variant Annotation
MEDLINE was searched systematically through April 30, 2017, for all case reports of patients with aHUS with identifiable genetic variants in any of the 10 genes known to be associated with aHUS: C3, CD46/MCP, CFB, CFH, CFHR1, CFHR3, CFHR5, CFI, DGKE, and THBD.3,19 If reported, information about the patients’ relatives was also recorded. When possible, genetic variants were annotated with patient demographics (e.g., genetic ancestry, age, sex), as well as specific disease characteristics such as onset, severity, etc. All data were stored in a relational database to allow data retrieval using structured query language.
To account for repeated-case reporting in the literature, patients were first matched based on age, sex, onset, genetic ancestry, and variant information. Potential matches were manually checked before being merged. In cases where one or more of the fields (e.g., age, sex, onset, genetic ancestry) were missing, the original articles were double-checked to ensure that they referenced each other and came from the same source (i.e., authors). All matches had to have the same genetic variant(s). We designated the resulting cases the “aHUS literature cohort.”
Variant Pathogenicity Analysis (Enrichment Analysis)
For every variant (e.g., allele A) present in both the aHUS literature cohort and the gnomAD reference population,17 a two-by-two contingency table can be constructed:
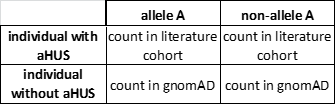
A right-tailed Fisher exact test was performed to evaluate whether allele A was enriched in the aHUS literature cohort as compared with that in the reference population gnomAD (i.e., non-aHUS population). P values were Bonferroni-corrected to account for multiple tests performed.
The same analysis was performed comparing the variants’ AF in the non-Finnish European subgroup (63 369 subjects) from gnomAD and that of patients of non-Finnish European ancestry (1261 patients) in the literature cohort.
In Silico Pathogenicity Prediction
The pathogenicity of genetic variants was evaluated using four of the representative methods included in the Variant Effect Predictor (VEP) tool20: Sorts Intolerant From Tolerant substitutions (SIFT),21 PROtein Variation Effect ANalyzer (PROVEAN),22 Functional Analysis Through Hidden Markov Models (FATHMM),23 and MutationTaster.24 Default settings were used to run the software. The results were then classified as either “deleterious” with a value of “1,” or “tolerated” with a value of “0” based on the specific criteria per method. For SIFT, the “deleterious” category included both predicted “deleterious” and “deleterious_low_confidence,” otherwise “tolerated”; for PROVEAN, the “deleterious” category included predicted “damaging,” otherwise “neutral” and in case of multiple predictions, more “damaging” results were required to be designated as “deleterious”; for FATHMM, the “deleterious” category included predicted “damaging,” otherwise “tolerated,” and in case of multiple predictions, more “damaging” results were required to be designated as “deleterious”; for MutationTaster, the “deleterious” category included both predicted “disease_causing_automatic” (A) and “disease_causing” (D), otherwise “tolerated,” and in case of multiple predictions, more A/D results were required to be designated as “deleterious.” In the end, the numeric values of the predictions were summed; thus, the final value represented how many methods made “deleterious” predictions.
Adjustment of AF in Patients With aHUS
Before statistical analyses were performed, the AFs of the variants in the aHUS literature cohort were adjusted by a factor of two because this cohort only included patients with identifiable genetic variants, who account for approximately 50% of the total aHUS patient population.
Results
Genetic Variants in the aHUS Literature Cohort Have Similar Distributions as in Reported Patient Cohorts
A systematic mining of the literature identified 1652 patients with aHUS with mutations in at least one of the 10 complement genes that have been shown to be associated with the disease.3,19 This group of patients was designated as the aHUS literature cohort. Table 1 contains a breakdown of the patients in the literature cohort by mutated genes (see column “Observed % Patient”). Because the literature cohort had only patients with identifiable genetic variants, who account for approximately 50% of the total aHUS patient population,3,19 the patient percentages were adjusted by a factor of two.
Table 1 also contains the breakdown of patients by mutated genes from published aHUS cohort studies (see column “Expected % Patient”).19 It has been shown that CFH is the most frequently mutated gene in these patients, accounting for approximately 20% to 30% of the genetic predisposition to aHUS. Consistently, CFH was also the most-mutated gene in the literature cohort, with 733 cases, or approximately 22% of patients having one or more variant in this gene. Genetic variants observed in our cohort, such as C3 (7%), CD46/MCP (8%), CFB (2%), and CFI (6%), also showed similar mutation rates as that reported for the literature cohort. DGKE mutations have been identified in 5% to 27% of patients aged 1 year and younger.25,26 In the literature cohort, there were 136 patients with a disease onset at or before 12 months. Twenty-four or 8.8% of these young patients had at least one DKGE variant. Only one gene, THBD, had a lower mutation rate in the literature cohort compared with its published mutation rates in patients with aHUS.
Table 1. Summary of genetic variants identified in aHUS literature cohort
| Gene | Patient Number | Observed % Patient a | Expected % Patient b | Unique Variant c | Variant in gnomAD |
|---|---|---|---|---|---|
| CFH | 733 | 22.2 | 20-30 | 184 | 48 |
| CFI | 188 | 5.7 | 4-10 | 56 | 40 |
| CD46 | 257 | 7.8 | 5-15 | 73 | 22 |
| C3 | 234 | 7.1 | 2-10 | 58 | 17 |
| CFB | 65 | 2.0 | 1-4 | 21 | 3 |
| DGKE | 36 | 1.1 | 28 | 4 | |
| THBD | 41 | 1.2 | 3-5 | 12 | 9 |
| CFHR1 | 240 | 7.3 | 1 | 0 | |
| CFHR3 | 181 | 5.5 | 1 | 1 | |
| CFHR5 | 23 | 0.7 | 13 | 2 | |
| Total | 1652 d | 447 | 146 |
a Adjusted by a factor of 2 to account for the fact that this cohort includes patients with identifiable genetic variants.
b Loirat C, Frémeaux-Bacchi V. 2011. Orphanet J Rare Dis, 6:60; Noris M, Remuzzi G. 2015. Am J Kidney Dis, 66(2):359-75.
c Excluding structural variants with no defined breakpoints.
d Total number of patients - some patients have variants in two or more genes.
CD46/MCP, membrane cofactor protein; CFB, complement factor B; CFH, complement factor H; CFI, complement factor I; CFHR1, complement factor H-related protein 1; CFHR3, complement factor H-related protein 3; CFHR5, complement factor H-related protein 5; DGKE, diacylglycerol kinase epsilon; THBD, thrombomodulin.
Most patients with aHUS have a mutation in only one gene and carry only one variant (Figure 1A and 1B). Among those patients who carry multiple genetic variants, the CFH, CFHR1-5 gene cluster on chromosome 1 is usually involved. The high sequence homology among these genes predisposes this genomic region to gene conversions and genomic rearrangements.27,28,29 Indeed, most of the variants in this region are structural variants with no defined breakpoints.
Consistent with the fact that aHUS is a rare disease, the majority of the variants collected were extremely rare. All but four of them had an AF ≤ 1% (Figure 1C). In the population database gnomAD,17 which has sequence data for more than 138,000 individuals, 301 of the variants are not present (Figure 1C). According to ACMG guidelines, these 301 variants have moderate evidence for pathogenicity (PM2).14 Given that the gene mutation rates in the literature cohort were in good agreement with those reported in the literature, we concluded that we could use this cohort to estimate AF in the aHUS population for a variant of interest.
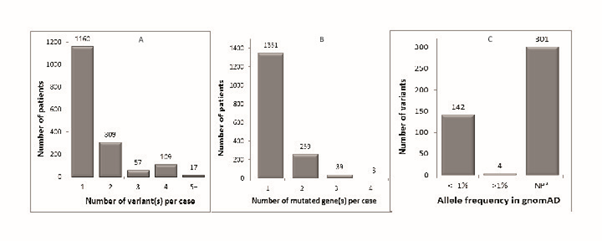
Figure 1: Distribution of genetic variants in aHUS literature cohort.
A. Numbers on bars are the patient numbers harboring the corresponding number of variant(s).
B. Numbers on bars are the patient numbers harboring variant(s) in the corresponding number of gene(s).
C. Numbers on bars are the variant numbers with the corresponding allele frequency. The allele frequency is based on gnomAD. NP: not present.
aHUS, atypical hemolytic uremic syndrome; gnomAD, Genome Aggregation Database; NP, not present.
Potentially Pathogenic Genetic Variants Were Identified by Enrichment Analysis
Although an identified variant in a causing gene can be suggestive of its pathogenicity, additional evidence such as functional/computational analysis is necessary to confirm the initial hypothesis. One approach is to examine whether such a genetic variant is statistically enriched in the patient population compared with a reference population. We employed this approach to evaluate the variants collected from a patient with aHUS and used the literature cohort and gnomAD17 as the patient and reference populations, respectively.
For each of the 146 variants present in both the literature cohort and gnomAD (Figure 1C), a Fisher exact test was utilized to compare the AFs in both groups. Using a cutoff of Bonferroni-corrected P value of less than or equal to 0.05, 40 variants were found to be significantly enriched in the literature patient cohort (Table S1). An examination of the AFs in gnomAD showed that all of the 40 variants are extremely rare (AF ≤ 0.001). Clearly, enrichment analysis can be applied to all types of genetic variants; among the 40 variants statistically enriched in patients, there were 27 missense, six stop gained, four splice, and two frameshift variants and one insertion/deletion variant.
We also ran the analysis by matching the genetic ancestry of patient and reference populations. Because non-Finnish European was the only ethnicity well represented in the literature cohort (1261 patients) and gnomAD (63 369 individuals), the analysis was performed using this subpopulation. There were 100 variants remaining when only non-Finnish Europeans were considered: 34 were found to be overrepresented in patients with aHUS. Figure 2 depicts the overlaps of the two analysis results: 33 variants were found to be enriched in patients regardless of ethnicity; only one variant was found to be enriched in patients when only non-Finnish Europeans were considered; seven variants were found to be enriched in patients when all ethnicities were included. However, three of the seven variants were due to the fact that they were missing in non-Finnish European populations (Figure 2 and Table S1).
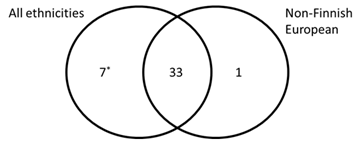
Figure 2: Comparison of enrichment analysis results using all ethnicities or non-Finnish European.
The venn diagram represents the overlap of variants exhibiting significant enrichment in aHUS patients when all ethnicities (left) or non-Finnish European (right) was considered. gnomeAD was used as reference population control.
*Three of 7 variants are not present in the non-Finnish European group in the Genome Aggregation Database.
In Silico Pathogenic Prediction
Numerous computational methods have been developed to predict the functional impact of genetic variant at the protein level. We used four of the representative methods (SIFT, PROVEAN, FATHMM, and MutationTaster) included in the VEP tool to evaluate the pathogenicity of the genetic variants identified in patients with aHUS. Two of the methods, SIFT21 and PROVEAN,22 are primarily based on amino acid sequence homology and protein structure information. FATHMM23 utilizes hidden Markov models weighted for human mutations, whereas MutationTaster uses a Bayes classifier to assess the results from a battery of in silico analyses and determine the combined effect on protein function.24
The complete prediction results are presented in Table S2. There was no output for variants such as insertions, deletions, frameshifts, or those affecting the splicing regions. In cases of gained stop codons, only MutationTaster could predict their impacts, but there were exceptions. In two cases where the mutations caused a frameshift before a stop codon, MutationTaster could no longer make a prediction. In addition, MutationTaster failed to predict a gained stop codon at amino acid position 160 in CFHR1; this mutation is right in the middle of the protein and will lead to a large truncation of the protein. Finally, only SIFT could correctly predict the deleterious effect of the loss of a start codon.
The summary of the prediction results of 282 missense mutations is shown in Table S3. Sixty-six or approximately 23% of these variants were not predicted as “deleterious” by any of the methods. By contrast, 43 variants were predicted to be “deleterious” by all four methods. Interestingly, a little more than half of the variants (22 of 43) involve the substitution of a cysteine residue by another amino acid (Table S2). Because cysteine residue is sometimes involved in forming disulfide bonds, which play an important role in the folding and stability of proteins, its substitution can be deleterious for maintaining protein structure and function. For example, CFH contains 20 cysteine-rich short consensus repeats. These repeats create a beta-sandwich arrangement held together by disulfide bonds formed by cysteine residues.30,31
The concordance of missense variant prediction varied among the methods utilized (Table S4). The two homology-based methods, SIFT and PROVEAN, shared high concordance in the prediction results. FATHMM predicted the fewest number of “deleterious” variants, and its output was least consistent with those of the other methods.
Concordance Between Prediction and Enrichment Analyses
The concordance of results generated by computational prediction and enrichment analyses is shown in Table S5. In general, if a variant was significantly enriched in the patient population, it was more likely to have a “deleterious” prediction by at least one of the prediction methods. Conversely, if a variant was predicted to be “deleterious,” it was more likely to be statistically significantly enriched in the patient population.
When using “enriched” variants (corrected P ≤ 0.05) as benchmarks for performance, FATHMM performed very well in terms of specificity (rows “Specificity – prediction”). However, the homology-based methods SIFT and PROVEAN were more sensitive, especially for missense variants, likely because these methods utilize protein sequence alignments in their predictions (rows “Sensitivity – prediction”). Overall, SIFT and PROVEAN have the better combination of sensitivity and specificity compared with the other prediction methods.
Conversely, when using “deleterious” variants (predicted by at least one tool) as benchmarks to assess the performance of enrichment analysis, the best concordance was with MutationTaster when all variants are considered. However, when only missense variants were included, results from the enrichment analysis also had very good concordance with those generated by SIFT and PROVEAN (rows “Specificity – enrichment” and “Sensitivity – enrichment”, Table S5).
An enrichment analysis can evaluate variants that are completely missed by prediction methods. For example, most of the variants flagged as potentially deleterious by only the enrichment analysis are variants in the splicing regions, deletions, and frameshifts (Table S6).
To examine the discordance between computational prediction and enrichment analyses, we used a scheme as depicted in Figure 3 to identify variants of discordance. To minimize random sampling bias, we examined variants with at least five allele counts in the literature cohort. The scheme identified five variants that were predicted by at least one computational method to be “deleterious” but exhibited no enrichment in the patient population. One of these variants (CFH c.2850G>T, p.Q950H) is within the SCR16 repeat in the CFH protein and is highly conserved among vertebrates (Figure 4A).32 As expected, SIFT and PROVEAN predicted it to be deleterious. This variant appears to predispose a person to develop aHUS33 but has incomplete penetrance.34,35 There was only one variant not predicted to be “deleterious” but showed enrichment in the literature cohort (Figure 3). CFH c.481G>T p.Ala161Ser showed enrichment regardless of whether the non-Finnish European subgroup was considered. This variant is located in SCR3 and affects the cofactor activity of CFH.36 This position exhibits moderate conservation among vertebrates as shown in Figure 4B.
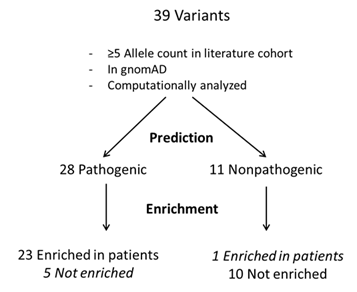
Figure 3: Concordance between prediction and enrichment analyses of variant pathogenicity.
Flowchart showing variants that passed the specified criteria.
gnomAD, Genome Aggregation Database.

Figure 4: Examples of variants showing discordance between in silico prediction and enrichment analysis.
Multiple sequence alignments were taken from Homologene.32 Representative variants predicted to be “deleterious” by one approach (e.g. in silico prediction) but not the other (e.g. enrichment analysis) were examined to understand possible reason(s) of discordance between the two approaches.
Discussion
The biggest challenge in diagnosing and treating any rare disease is, arguably, a lack of information. In the past two decades, the development of new technologies, such as the next-generation sequencing methods, has greatly increased the number of rare diseases with an identified genetic cause. However, interpreting the large amount of genetic data is challenging, and many genetic variants identified in patients remain uncharacterized and their significance to disease phenotypes unknown.14 Improved understanding of the genotype-to-phenotype correlation is important to guide long-term treatment and management decisions for these patients.
We systematically explored the case studies of aHUS in MEDLINE and created a literature cohort of more than 1600 patients with aHUS. This cohort was much larger than any published patient cohort of this disease and allowed for additional characterization of rare genetic variants using computational approaches. Importantly, the gene and variant distributions in this virtual cohort were in good agreement with what has been reported in actual cohorts of patients with aHUS (Table 1).
We analyzed the pathogenicity of genetic variants identified in 10 genes associated with aHUS. A Fisher exact test was utilized to compare those variants’ AFs in the literature cohort with those in a reference population, gnomAD.17 Even though gnomAD contains sequencing data of more than 138 000 individuals, most of the identified variants (301 of 447) are not present, highlighting the challenge and importance of obtaining more sequencing data from individuals of diverse genetic ancestry to enable assessment of rare and ultra-rare variants. Based on the current ACMG guidelines, the 301 variants have moderate evidence for pathogenicity (PM2).
Enrichment analysis enabled by a large patient cohort can provide novel insight into the pathogenicity of variants. Our enrichment analysis provides strong evidence for pathogenicity (PS4) based on ACMG guidelines. In our analysis, 40 variants showed enrichment in the literature cohort as compared with the reference population. 22 of these variants have been analyzed using functional assays and all 22 were found to be either “pathogenic” or “probably pathogenic” (Table S1).37-43
Moreover, the enrichment method can be applied to variant types that cannot be processed by the prediction methods. For example, seven out of the ten variants enriched in the patient population but not flagged as “deleterious” by any of the prediction methods are insertion/deletion, frameshift, splice, and stop codon variants (Table S6). This shows the importance of enrichment analysis because at least 30% of all identified variants are not missense variants and cannot be processed by many prediction methods.
The ten variants also include three pathogenic missense variants occurring at amino acid positions not conserved evolutionarily (Table S6). The consequence of the mutations can be appreciated by inspecting the crystal structures of complement complexes. For example, CFH variants c.481G>T, p.Ala161Ser and c.2908A>G, p.Ile970Val are located in SCR3 and SCR16, which mediate the binding of CFH to C3b and C3d,36,44 respectively. Nevertheless, neither of the prediction tools can take this information into account.
We also ran the enrichment analysis by matching genetic ancestry of the literature cohort and the reference population. Because only the non-Finnish European population had sufficient numbers, the analysis was done only for this subgroup. The concordance between the analyses (when all groups or when only non-Finnish Europeans were included) was very high: 33 variants were found to be enriched in both analyses (Figure 2). For the four variants found to be enriched only when all ethnicities were considered, higher allele counts in non-Finnish European patients were observed, but the difference was not statistically significant. The lack of statistical significance might be due to the lack of power because of reduced population numbers. There was only one variant, CFI c.1270A>C p.Ile424Leu, found to be enriched only when non-Finnish Europeans were considered. This variant showed clear enrichment in non-Finnish Europeans. However, it was also present at high AFs in other populations, such as Africans (285 of 24 024, or approximately 0.012), raising the doubt whether the variant is truly pathogenic.
We recognize that there are caveats in creating patient cohorts from the literature and computational analyses. First, genes tested are not likely to be uniform across all literature cases. Differences in testing panels, sequencing, and analyzing methods likely will lead to heterogeneity in data collection. For example, if a gene was not tested, it could potentially lead to false negatives (e.g., variant exists, but not identified). Second, although we adjusted the AF in patients by a factor of two, this is still an unknown factor and may be different from population to population. Third, enrichment and prediction methods may not be sensitive for variants with incomplete penetrance and variants acting in combinations. Additional information such as genetic segregation data is needed to supplement the computational methods. Lastly, many structural variants remain a challenge to be assessed by enrichment analysis because of undefined breakpoints. If possible, these variants should be categorized at the gene level or by a specific focused region (e.g., missing a particular exon), so that AF can be calculated and the analysis performed.
Conclusions
We have presented an in silico framework where an artificial patient cohort was created and used to assess the pathogenicity of genetic variants. This framework can be efficiently applied to rare diseases where information is sparse to shed light on genotype-phenotype associations and ultimately help diagnose and treat patients with rare diseases who are in desperate need of better health care.
Acknowledgments
The authors would like to acknowledge Guillermo del Angel and John Reynders of Alexion Pharmaceuticals, Inc., for critical review of the manuscript, and Guillermo del Angel for database querying and formatting. The authors would also like to acknowledge Peloton Advantage, LLC, which provided medical writing/editorial/layout support with funding from Alexion Pharmaceuticals, Inc.
Conflict of Interests
RRJ is an employee of Alexion Pharmaceuticals, INC. and owns the company stocks.
Funding Information
Alexion Pharmaceuticals, Inc. provided funding to this work.
Author’s contributions
RRJ did the analyses and wrote the manuscript. TS and NK directed the literature mining efforts. All authors read and approved the final manuscript.
References
-
- Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847-2856.
- Constantinescu AR, Bitzan M, Weiss LS, et al. Non-enteropathic hemolytic uremic syndrome: causes and short-term course. Am J Kidney Dis. 2004;43(6):976-982.
- Loirat C, Fremeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis. 2011;6:60.
- Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2007;18(8):2392-2400.
- Rafiq A, Tariq H, Abbas N, Shenoy R. Atypical hemolytic-uremic syndrome: a case report and literature review. Am J Case Rep. 2015;16:109-114.
- Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(17):1676-1687.
- Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8(4):554-562.
- Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844-1859.
- Campistol JM, Arias M, Ariceta G, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia. 2015;35(5):421-447.
- Goodship TH, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Int. 2017;91(3):539-551.
- Loirat C, Fakhouri F, Ariceta G, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2016;31(1):15-39.
- Noris M, Remuzzi G. Cardiovascular complications in atypical haemolytic uraemic syndrome. Nat Rev Nephrol. 2014;10(3):174-180.
- Bu F, Maga T, Meyer NC, et al. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2014;25(1):55-64.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424.
- Rodriguez de Cordoba S, Hidalgo MS, Pinto S, Tortajada A. Genetics of atypical hemolytic uremic syndrome (aHUS). Semin Thromb Hemost. 2014;40(4):422-430.
- Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6(2):95-108.
- Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285-291.
- Genome aggregation database. 2017. http://gnomad.broadinstitute.org/. Accessed: December 21, 2017.
- Noris M, Remuzzi G. Glomerular diseases dependent on complement activation, including atypical hemolytic uremic syndrome, membranoproliferative glomerulonephritis, and C3 glomerulopathy: core curriculum 2015. Am J Kidney Dis. 2015;66(2):359-375.
- Variant effect predictor. 2017. https://www.ensembl.org/info/docs/tools/index.html. Accessed: December 18, 2017.
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863-874.
- Choi Y, Sims GE, Murphy S, Miller JR, et al. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7(10):e46688.
- Shihab HA, Gough J, Cooper DN, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34(1):57-65.
- Schwarz JM, Rodelsperger C, Schuelke M, et al. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575-576.
- Sanchez Chinchilla D, Pinto S, Hoppe B, et al. Complement mutations in diacylglycerol kinase-epsilon-associated atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2014;9(9):1611-1619.
- Lemaire M, Fremeaux-Bacchi V, Schaefer F, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet. 2013;45(5):531-536.
- Diaz-Guillen MA, Rodriguez de Cordoba S, Heine-Suner D. A radiation hybrid map of complement factor H and factor H-related genes. Immunogenetics. 1999;49(6):549-552.
- Male DA, Ormsby RJ, Ranganathan S, et al. Complement factor H: sequence analysis of 221 kb of human genomic DNA containing the entire fH, fHR-1 and fHR-3 genes. Mol Immunol. 2000;37(1-2):41-52.
- Nester CM, Barbour T, de Cordoba SR, et al. Atypical aHUS: State of the art. Mol Immunol. 2015; 67(1):31-42.
- Cho H. Complement regulation: physiology and disease relevance. Korean J Pediatr. 2015;58(7):239-244.
- Rodriguez de Cordoba S, Esparza-Gordillo J, Goicoechea de Jorge E, et al. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol. 2004;41(4):355-367.
- HomoloGene. 2017. https://www.ncbi.nlm.nih.gov/homologene. Accessed: June 12, 2017.
- Sartz L, Olin AI, Kristoffersson AC, et al. A novel C3 mutation causing increased formation of the C3 convertase in familial atypical hemolytic uremic syndrome. J Immunol. 2012;188(4):2030-2037.
- Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267-1279.
- Kavanagh D, Goodship T. Genetics and complement in atypical HUS. Pediatr Nephrol. 2010;25(12):2431-2442.
- Fakhouri F, Roumenina L, Provot F, et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol. 2010;21e(5):859-867.
- Schramm EC, Roumenina LT, Rybkine T, et al. Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood. 2015;125(15):2359–2369.
- Merinero HM, García SP, García-Fernández J, et al. Complete functional characterization of disease-associated genetic variants in the complement factor H gene. Kidney Int. 2018;93(2):470-481.
- Roumenina LT, Roquigny R, Blanc C, et al. Functional evaluation of factor H genetic and acquired abnormalities: application for atypical hemolytic uremic syndrome (aHUS). Methods Mol Biol. 2014;1100:237-47.
- Delvaeye M, Noris M, De Vriese A, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(4):345-57.
- Kavanagh D, Yu Y, Schramm EC, et al. Rare genetic variants in the CFI gene are associated with advanced age-related macular degeneration and commonly result in reduced serum factor I levels. Hum Mol Genet. 2015 Jul 1;24(13):3861-70.
- Marinozzi MC, Vergoz L, Rybkine T, et al. Complement factor B mutations in atypical hemolytic uremic syndrome-disease-relevant or benign? J Am Soc Nephrol. 2014;25(9):2053-65.
- Liszewski MK, Atkinson JP. Complement regulator CD46: genetic variants and disease associations. Hum Genomics. 2015;9:7.
- Saunders RE, Abarrategui-Garrido C, Frémeaux-Bacchi V, et al. The interactive Factor H-atypical hemolytic uremic syndrome mutation database and website: update and integration of membrane cofactor protein and Factor I mutations with structural models. Hum Mutat. 2007;28(3):222-34.