
IgG-4 related disease: A mini-review
Ramy Sedhom1*, Daniel Sedhom1 and Roger Strair2
1Department of Internal Medicine, Rutgers Robert Wood Johnson Medical School, New Brunswick, New Jersey, USA
2Division of Hematology and Oncology, Rutgers Cancer Institute of New Jersey, New Brunswick, New Jersey, USA
Abstract
IgG4-related disease is a fibroinflammatory condition that mimics many malignant, infectious, and inflammatory disorders. Histopathology is key to diagnosis and is hallmarked by tumor like infiltration of IgG4 positive plasma cells in tissues, with dense lymphoplasmacytic infiltrate, storiform fibrosis and oblierative phlebitis. Disease has been reported in virtually every organ system1-4. Though the underlying pathophysiology is still unclear, untreated disease ultimately leads to irreversible fibrosis. We describe the pathogenesis, diagnosis, and relevant features of IgG4-related disease and discuss current evidence regarding treatment.
The role of IgG4 in disease
IgG4 is an antibody with unique structure and function5. It is the least abundant IgG subclass, accounting for <5% of total IgG. IgG4 is a noninflammatory immunoglobulin that might function in antigen-capture, preventing the binding of yet unidentified antigens that drive inflammatory processes. Responses of IgG4 have been induced by prolonged or repeated antigen exposures (6). IgG4 production is thought to be controlled primarily by type 2 helper T (Th2) cells5,6.
The molecule of IgG4 is thought to play an important role in immune mediated conditions. IgG4 antibodies against desmoglein are responsible for the formation of cutaneous blisters in pemphigus vulgaris7. They also have been implicated in idiopathic membranous glomerulonephritis and in thrombotic thrombocytopenic purpura8,9.
Pathophysiology
The pathogenesis of IgG4-related disease is vague and still immature in scientific study. The two prevailing theories underlying observed pathology include 1) induction of a polarized CD4+ T cell that activates innate immune cells responsible for the development of fibrosis and 2) a negative feedback process involving generation of IgG4 secreting plasmablasts, plasma cells, and IgG4 antibodies responsible for preventing autoimmunity. Autoimmunity has been hypothesized to be a potential initial immunologic stimulus for the Th2-cell response in IgG4 related disease10-12.
T cells are commonly linked in disease pathogenesis due to the observation that many CD4+ T cells are present at sites of inflammation in IgG4-related disease. Recent published studies have also shown that Th2 memory cells do aggregate in a large percentage of people with IgG4-related disease if they have concomitant atopy13,14. Large clonal expansions of cytotoxic CD4+ cytotoxic T lymphocytes have been found in affected tissues, in addition to activated CD19+CD20-CD38highCD27high B cells (called plasmablasts)14. The reason and nature of the disease-causing CD4+ T cells remains to be elucidated.
Eosinophilia and elevations in serum IgE levels have been found in 40% of patients with IgG4 related disease; extreme cases have been reported resembling eosinophilic organopathy13. Ultimately, an encounter with a microbe likely triggers tissue damage with breaks in immunologic tolerance. Activated CD4+ T-helper cells induce a fibrosis driven cytokine pathway. These memory T cells that help coordinate disease are presumably sustained by plasmablasts, explaining potential responses to B-cell depletion15.
Pathologic features of IgG4-related disease
Histologic examination of biopsy specimens is the gold standard in diagnosing IgG4-related disease. Even with supportive histopathology, clinical symptoms and serologic findings are needed to confirm diagnosis. Serum IgG4 is elevated in most cases, but about 30% of histologically confirmed cases have normal levels of IgG4, which can lead to false negatives3,16,17. In general, the specificity and positive predictive value of serum IgG4 are low. Misdiagnosis is common due to overreliance on mild elevations of serum IgG4 or the presence of IgG4-positive plasma cells in tissue (Table 1).
| The histopathological findings include: |
| • A dense lymphoplasmacytic infiltrate, |
| • Storiform fibrosis, and |
| • Obliterative phlebitis |
| The presence of these findings, with or without tissue eosinophilia, is strongly suggestive of IgG4-related disease. Disease is more suggestive if accompanied by increased numbers of IgG4-positive plasma cells. Sizeable minority have normal serum IgG4 despite classic histopathological changes in tissue |
| The diagnosis cannot be made solely by the number of IgG4-positive plasma cells as many other disease states have similar findings. |
| Serum IgG4 concentrations are neither sensitive nor specific for this disease. Histologic confirmation of the diagnosis by biopsy of an involved organ is necessary. |
Table 1. Diagnostic criteria for IgG4-Related Disease
The typical histologic abnormalities are a dense lymphoplasmacytic infiltrate, storiform fibrosis, and obliterative phlebitis18. Due to patchy distribution, storiform fibrosis can escape detection due to sampling error, especially if obtained by needle biopsy (Figure 1). Fibrosis is very rare in lymph nodes; diagnosis of IgG4-related disease is therefore extremely difficult on lymph-node pathology alone. Granulomas are unusual and if found suggest alternative etiology. Though histologic features are similar for all organs, subtle pathologic variations exist among affected organ systems. Immunohistochemical confirmation with IgG4 immunostaining is required (Figure 2).
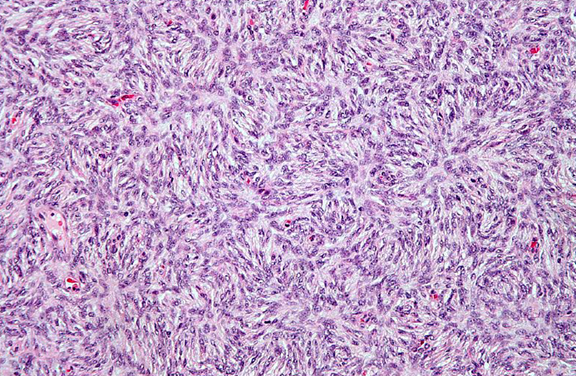
Figure 1. Fibrosis has a characteristic “storiform” pattern, typified by a cartwheel appearance of the arranged fibroblasts and inflammatory cells.
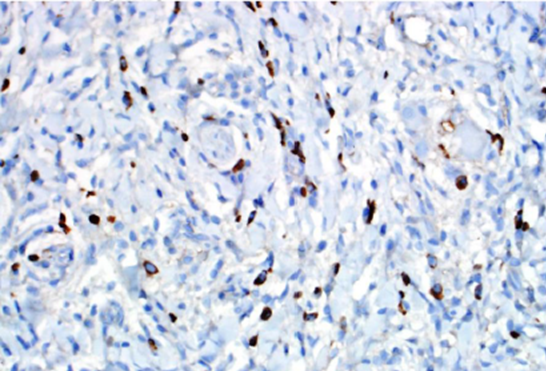
Figure 2. IgG4 immunohistochemical stain highlights many IgG4-positive plasma cells within this single high-powered field.
Pathologic features of IgG4-related disease
Inflammatory infiltrates are composed of both T and B-lymphocytes. High numbers of IgG4 positive plasma cells are required for diagnosis, but are commonly found in other inflammatory infiltrates such as xanthogranulomatous disease, granulomatosis with polyangitis, Castelman’s disease, sarcoidosis and neoplastic disorders. Lymphoma is the closest histologic relative of IgG4 related disease and clonal studies should be considered to rule out malignancy. Simple presence of IgG4 positive plasma cells is insufficient to establish a diagnosis of IgG4-related disease. A variety of cutoff points for the number if IgG4-positive plasma cells required for diagnosis have been suggested, ranging from 10 to 50 IgG4 positive plasma cells per high-power field19-21. In addition, the ratio of IgG4/IgG positive plasma cells in tissues should be greater than 40%. Diagnosis is important to consider early in disease, as late stage is hallmarked by fibrosis and unlikely to respond to treatment. Several sets of diagnostic criteria have been devised and proposed for practical use by nonspecialists22-25.
Epidemiologic characteristics
Epidemiologic understanding of IgG4-related disease is limited due to insufficient awareness of the diagnosis and its only recent appearance in the medical literature. Several observations however have been noted: patients are mostly male and older than 50 years of age26. Male predominance is much more prevalent in disease involving the kidney and retroperitoneum, with reported prevalence as high as 90%27. The higher disease prevalence in men is in contrast to other autoimmune diseases hallmarked by female predominance by nine to one. The differences in expression between the two sexes are unclear.
The role of imaging
Although definitive diagnosis of IgG4-related disease requires histopathology analysis, radiologic imaging plays an important role in demonstrating features suggestive of the diagnosis. Imaging workup for IgG4-related disease should include a CT scan of the chest, abdomen and pelvis to detect possible multiorgan involvement, specifics of which will be further described. Cross sectional imaging can supplement diagnostic criteria for IgG4-related disease. Key features can be seen based on the involved organ. In general, imaging studies will demonstrate infiltration and enlargement of involved organs. Other general radiologic features include glandular swelling, nodularity, organ wall thickening, fibrosis, and lymphadenopathy. Familiarity with radiologic findings can help avoid delays in diagnosis, unnecessary surgical procedures, establish a biopsy site for histologic analysis, and allow for timely initiation of appropriate therapy. Use of positron emission tomography-computed tomography (PET-CT) is advocated by many. Studies have shown its utility for diagnosis, staging, and monitoring of response. This imaging method, in comparison to ultrasonography and CT has yielded better results28.
The role of imaging
The clinical features of IgG4-related disease (Table 2) will be discussed in the context of the body cavities in which they occur and typical radiographic findings:
| • Orbits & periorbital tissues: tumefactive lesions, orbital pseudotumor, dacrocystitis, orbital myositis
• Ears, nose, sinuses: allergic phenomena, eosinophilic angiocentric fibrosis • Salivary glands: submanidublar and/or parotid enlargement• Meninges: predilection for dura, formation of mass lesions • Lymph nodes: lymphadenopathy• Thyroid: Riedel’s thyroiditis (fibrosing variant of Hashimoto’s thyroiditis) • Lungs: inflammatory pseudotumor, central airway disease, diffuse interstitial pneumonia, pleuritis• Aorta: lymphoplasmacytic aortitis, periaortitis, periarteritis, inflammatory aneurysms • Retroperitoneum: retroperitoneal fibrosis• Kidneys: tubulointerstitial nephritis, asymptomatic tumoral lesions • Pancreas: type I autoimmune pancreatitis• Biliary tree: sclerosing cholangitis • Liver: inflammatory mass lesions (pseudotumor)• Gallbladder: lymphoplasmacytic sclerosing cholecystitis • Sclerosing mediastinitis and mesenteritis• Breast, prostate, skin, and peripheral nerve involvement also described |
Table 2. Organ Involvement in IgG4-Related Diseases (IgG4-RD)
Constitutional and musculoskeletal symptoms
The presentation of IgG4-related disease is typically subacute. Symptoms may wax and wane with spontaneous improvement, with years of disease inactivity. Fatigue and musculoskeletal complaints are common, especially with multiorgan system involvement. Constitutional features, such as fevers and elevations of inflammatory markers, are not typical.
Head and neck
Salivary glands may be affected by IgG4-related disease. Submandibular gland involvement is particularly characteristic, and carries the eponym Kuttner’s tumor29. In contrast to Sjogren’s syndrome, symptoms improve with immunosuppression. Isolated enlargement of the submandibular gland, common to IgG4-related disease, is uncommon in Sjogren’s disease, which is hallmarked by parotid gland enlargement. CT imaging demonstrates homogenous attenuation and enhancement. On MRI, low signal intensity is seen on T2 imaging secondary to fibrosis30.
Allergic features are most prominent in the ears, nose, and throat. Presentation includes allergic rhinitis, nasal polyps, chronic sinusitis, nasal obstruction, and rhinorrhea. Mild to moderate peripheral eosinophilia and elevated serum IgE levels are common. Despite these findings, most patients with IgG4-related disease lack atopy31.
IgG4-related disease can lead to inflammation in the pharynx, hypopharynx, and can present with mass lesions32. Mass lesions can occur in the sinuses, middle ear, and facial bones. Tracheal inflammation and vocal cord involvement have been described, but further studies are needed to better elucidate its relationship to subglottic stenosis33.
Thyroid gland enlargement secondary to Riedel’s thyroiditis can present with neck pain, dyspnea, dysphagia and dysphonia. They thyroid gland can become sclerotic overtime and disease extension to adjacent tissues is well documented. CT demonstrates focal or diffuse low attenuation of the thyroid34.
Intracranial disease
Intracranial presentations of pachymeningitis and hypophysitis have been noted. Presentation includes headache, radiculopathy, cranial nerve palsies, or symptoms consistent with spinal cord compression. Mass lesions have also been seen, leading to the disease description of hypertrophic patchymeningitis35. Of note, IgG4-related disease does not typically affect brain parenchyma.
Lungs
Lung manifestations of IgG4-related disease are diverse. The disease tracks along bronchi and blood vessels, and can be seen on imaging as pulmonary nodules, ground-glass opacities, pleural thickening or interstitial lung disease36. Clinical symptoms include cough, hemoptysis, dyspnea, pleural effusions, and chest pain. More vascular lesions and obliterative phlebitis occur in the lungs (Figure 3). In addition, neutrophilic infiltration is more common in the lungs than in other organs36,37. Pleural effusions can occur. On CT, findings are nonspecific, and usually appear as ground glass opacities. Honeycombing may also be seen, making it difficult to distinguish from nonspecific interstitial pneumonia37.
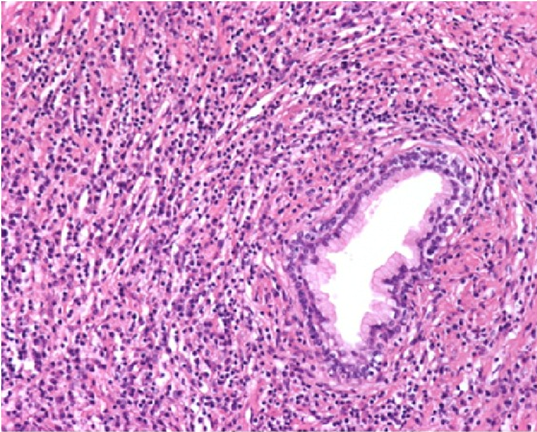
Figure 3. Dense inflammatory infiltrate with numerous plasma cells. Vessels show obliterative phlebitis.
Thoracic aorta
IgG4-releated aortitis can cause aneurysms or dissections of the thoracic aorta. Arterial wall thickening and aortic dilatation can be detected on cross imaging studies37. Coronary artery lesions are rare. Fibrosing mediastinitis has been described and can be difficult to treat as compression of mediastinal structures is not uncommon. Storiform fibrosis can be seen under the microscope38.
Pancreas
The pancreas was the first organ recognized as related to IgG4 disease4. Type 1 autoimmune pancreatitis presents with mild abdominal pain, weight loss, and painless and often obstructive jaundice. It is characterized histopathologically by lymphoplasmacystic sclerosing pancreatitis. Two main recognized patterns have been recognized: diffuse and focal4. Diffuse disease is more common and is characterized by an enlarged pancreas with absence of pancreatic clefts. Focal disease has a characteristic enlargement of the pancreatic head and appears very similar to mass like lesions. International diagnostic criteria have been proposed and include imaging, serum IgG4 concentrations, pancreatic histology, extrapancreatic disease, and glucocorticoid responsiveness22. Endoscopic ultrasound should be performed, with fine needle aspiration, to exclude pancreatic malignancy.
Biliary tract and liver
IgG4-related sclerosing cholangitis is commonly associated with type I autoimmune pancreatitis39. If untreated, it can progress to end-stage liver disease and is extremely difficult to differentiate from primary sclerosing cholangitis. The histology includes obliterative phlebitis and transmural fibrosis. Dense infiltrates of IgG4-positive plasma cells and T cells can be seen. IgG4-related disease can similarly affect the liver. Patients present with mass lesions and clinically with obstructive jaundice. If the mass lesions occlude the bile ducts, presentation may resemble cholangiocarcinoma40. Imaging may show focal or diffuse bile duct thickening, often associated with stenosis or upstream dilatation. These findings may similarly be depicted on ERCP or MRCP.
Retroperitoneal fibrosis & kidney involvement
A large percentage of idiopathic retroperitoneal fibrosis has been linked to IgG4-related disease41. Storiform fibrosis and obliterative phlebitis are commonly identified. In the kidney, tubulointerstitial disease is common. Although presentation is typically indolent, nephrotic syndrome and advanced renal failure can occur. Histopathology is similar to other organs42. Kidney disease is differentiated from other organs by profoundly low complement levels. At this time, the cause of low complement is poorly understood. Renal lesions are usually not visible on nonenhanced CT. MR imaging show low intensity on both T1 and T2 weighted images, with mild enhancement on T1 after contrast administration43.
Other organs
Many patients with IgG4-related disease have allergic features seen in atopy, eczema, chronic sinusitis and asthma. Modest elevations in eosinophils can be observed6. Cutaneous disease and skin manifestations are rare, with typical lesions being erythematous, flesh colored papules on the face44.
Serologic findings
Though the majority of patients with IgG4-related disease have elevations in serum IgG4 levels, 30% of patients do not, despite confirmation of tissue pathology11. In addition, data regarding serial measurements of IgG4 concentrations as indicators of disease activity are mixed45. IgG4 levels do decrease after initiation of glucocorticoid treatment in the majority of patients, but regularly remain above baseline weeks to months later11. Identifying high numbers of circulating plasmablasts by flow cytometry is more sensitive than serum IgG4 concentrations, but testing assays are not widely accessible46,47.
Treatment
No randomized trials exist at this time and large retrospective studies are few. Review of current literature suggests early aggressive treatment to prevent serious organ dysfunction and failure. Glucocorticoids are the first line of treatment. Unfortunately, many patients require a prolonged course. A consensus statement from Japan suggests starting with prednisolone at a dose of 0.6mg per kilogram for 2 to 4 weeks with a very slow taper, adjusted according to aggressiveness of disease48. Though glucocorticoids appear to be effective in the short-term, disease flairs and relapse are common despite maintenance therapy. Glucocorticoid sparing agents and DMARDS, such as azathioprine, mycophenolate mofetil, and methotrexate, have been used, though efficacy has not been validated in clinical trials. Rigorous assessment of these treatments is needed.
Patients with refractory disease appear to response to B-cell depletion with Rituximab, offering novel insight to the pathophysiology of this disorder15,49. Clinical response is rapid, with correlating decreases in IgG4 concentrations. B-cell depletion targets the subset of plasma cells that produce IgG4. This is likely achieved by depleting CD20+ B cells. Some authors have suggested the role of antigen presentation by IgG4+ plasmablasts prior to a cytokine cascade driving the fibrotic process14. These plasmablasts decline quickly after B-cell depletion and can be useful to know which patients should be individually targeted with Ritixumab therapy. Intravenous or subcutaneous immunoglobulin treatment has been successful in treating other inflammatory or immune mediated diseases, but has not yet been trialed in IgG4-related disease. Responsiveness to medical therapy is limited in late stage disease and in the case of extensive fibrosis, though case reports have been reported27.
Conclusions and future perspectives
IgG4-related disease is rare and underappreciated secondary to unawareness. Symptoms are variable and clinical presentation is reflective of affected organs. Early recognition and therapy are important to prevent serious and irreversible tissue damage. Further epidemiologic data are needed to confirm and validate early observations. Better understanding and knowledge of the dysregulation associated with IgG4-related disease will explain much about the immune system.
References
- Kamisawa T, Funata N, Hayashi Y, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003; 38: 982-4.
- Stone JH, Khosroshahi A, Hilgenberg A, et al. IgG4-related systemic disease and lymphoplasmacytic aortitis. Arthritis Rheum. 2009; 60: 3139-45.
- Saeki T, Saito A, Hiura T, et al. Lymphoplasmacytic infiltration of multiple organs with immunoreactivity for IgG4: IgG4-related systemic disease. Intern Med. 2006; 45: 163-7.
- Kamisawa T, Takuma K, Egawa N, et al. Autoimmune pancreatitis and IgG4-related sclerosing disease. Nat Rev Gastroenterol Hepatol. 2010; 7: 401-9.
- Nirula A, Glaser SM, Kalled SL, et al. A review of the biology of a unique immunoglobulin subtype. Curr Opin Rheumatol. 2011; 23: 119-24.
- Aalberse RC, Stapel SO, Schuurman J, et al. Immunoglobulin G4: an odd antibody. Clin Exp Allergy. 2009; 39: 469- 77.
- Rock B, Martins CR, Theofilopoulos AN, et al. The pathogenic effect of IgG4 autoantibodies in endemic pemphigus foliaceus fogo selvagem. N Engl J Med. 1989; 320: 1463-9.
- Beck LH Jr, Salant DJ. Membranous nephropathy: recent travels and new roads ahead. Kidney Int. 2010; 77: 765-70.
- Ferrari S, Mudde GC, Rieger M, et al. IgG subclass distribution of anti-ADAMTS13 antibodies in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost. 2009; 7: 1703-10.
- Aparisi L, Farre A, Gomez Cambronero L, et al. Antibodies to carbonic anhydrase and IgG4 levels in idiopathic chronic pancreatitis: relevance for diagnosis of autoimmune pancreatitis. Gut. 2005; 54: 703-9.
- Asada M, Nishio A, Uchida K, et al. Identification of a novel autoantibody against pancreatic secretory trypsin inhibitor in patients with autoimmune pancreatitis. Pancreas. 2006; 33: 20-6.
- Löhr JM, Faissner R, Koczan D, et al. Autoantibodies against the exocrine pancreas in autoimmune pancreatitis: gene and protein expression profiling and immunoassays identify pancreatic enzymes as a major target of the inflammatory process. Am J Gastroenterol. 2010; 105: 2060-71.
- Kanari H, Kagami S, Kashiwakuma D, et al. Role of Th2 cells in IgG4-related lacrimal gland enlargement. Int Arch Allergy Immunol. 2010; 152:Suppl 1:47-53.
- Mattoo H, Mahajan VS, Della Torre E, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014; 134: 679-87.
- Khosroshahi A, Carruthers M, Deshpande V, et al. Rituximab for the treatment of IgG4 related disease lessons from ten consecutive patients. Medicine. 2012; 91: 57-66.
- Strehl JD, Hartmann A, Agaimy A. Numerous IgG4 positive plasma cells are ubiquitous in diverse localised non-specific chronic inflammatory conditions and need to be distinguished from IgG4- related systemic disorders. J Clin Pathol. 2011; 64: 237-43.
- Sah RP, Chari ST. Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol. 2011; 23: 108-13.
- Zen Y, Nakanuma Y. IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol. 2010; 34: 1812-9.
- Dhall D, Suriawinata AA, Tang LH, et al. Use of immunohistochemistry for IgG4 in the distinction of autoimmune pancreatitis from peritumoral pancreatitis. Hum Pathol. 2010; 41: 643-52.
- Deshpande V, Chicano S, Finkelberg D, et al. Autoimmune pancreatitis: a systemic immune complex mediated disease. Am J Surg Pathol. 2006;30: 1537-45.
- Chari ST. Diagnosis of autoimmune pancreatitis using its five cardinal features: introducing the Mayo Clinic’s HISORt criteria. J Gastroenterol. 2007; 42: Suppl 18:39-41.
- Shimosegawa T, Chari ST, Frulloni L, et al. and the International Association of Pancreatology. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas. 2011; 40: 352–58.
- Ohara H, Okazaki K, Tsubouchi H, et al. and the Research Committee of IgG4-related Diseases and the Research Committee of Intractable Diseases of Liver and Biliary Tract and the Ministry of Health Labor and Welfare Japan and the Japan Biliary Association Clinical diagnostic criteria of IgG4-related sclerosing cholangitis 2012. J Hepatobiliary Pancreat Sci. 2012; 19: 536–42.
- Kawano M, Saeki T, Nakashima H, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011; 15: 615–26.
- Masaki Y, Sugai S, Umehara H. IgG4-related diseases including Mikulicz’s disease and sclerosing pancreatitis: diagnostic insights. J Rheumatol. 2010; 37: 1380–85.
- Raina A, Yadav D, Krasinskas AM, et al. Evaluation and management of autoimmune pancreatitis experience at a large US center. Am J Gastroenterol. 2009; 104: 2295-306.
- Zen Y, Onodera M, Inoue D, et al. Retroperitoneal fibrosis a clinicopathologic study with respect to immunoglobulin G4. Am J Surg Pathol. 2009; 33: 1833-9.
- Zhang J, Chen H, Ma Y, et al. Characterizing IgG4-related disease with FDG PET/CT: a prospective cohort study. Eur J Nucl Med Mol Imaging. 2014; 41:1624-34.
- Geyer JT, Ferry JA, Harris NL, et al. Chronic sclerosing sialadenitis (Kuttner tumor) is an IgG4-associated disease. Am J Surg Pathol. 2010; 34: 202–10.
- Fujita A, Sakai O, Chapman M, et al. IgG4-related disease of the head and neck CT and MR imaging manifestations. Radiographics. 2012; 32 (7): 1945-1958.
- Della Torre E, Mattoo H, Mahajan VS, et al. Prevalence of atopy eosinophilia and IgE elevation in IgG4-related disease. Allergy. 2014; 69: 269–72.
- Fatemi G, Fang MA. IgG4-related pharyngitis-an addition to the nomenclature of IgG4-related disease comment on the article by Stone et al. Arthritis Rheum. 2013; 65: 2217.
- Schiff AI, Gahl WA, Pittaluga S, et al. IgG4-related disease presenting as recurrent mastoiditis. Laryngoscope. 2012; 122: 681–84.
- Dahlgren M, Khosroshahi A, Nielsen GP, et al. Riedel’s thyroiditis and multifocal fibrosclerosis are part of the IgG4-related systemic disease spectrum. Arthritis Care Res. 2010; 62: 1312–18.
- Wallace ZS, Carruthers MN, Khosroshahi A, et al. IgG4-related disease and hypertrophic pachymeningitis as a common etiology. Medicine. 2013; 92: 206–16.
- Zen Y, Inoue D, Kitao A, et al. IgG4-related lung and pleural disease a clinicopathologic study of 21 cases. Am J Surg Pathol. 2009; 33: 1886–93.
- Kasashima S, Zen Y, Kawashima A, et al. A clinicopathologic study of immunoglobulin G4-related sclerosing disease of the thoracic aorta. J Vasc Surg. 2010; 52: 1587–95.
- Peikert T, Shrestha B, Aubry MC, et al. Histopathologic overlap between fibrosing mediastinitis and IgG4-related disease Int J Rheumatol. 2012; 207-56.
- Zen Y, Harada K, Sasaki M, et al. IgG4-related sclerosing cholangitis with and without hepatic inflammatory pseudotumor and sclerosing pancreatitis-associated sclerosing cholangitis do they belong to a spectrum of sclerosing pancreatitis. Am J Surg Pathol. 2004; 28: 1193–203.
- Mahajan V, Mattoo H, Deshpande V, et al. IgG4-Related Disease. Ann Rev Path. 2014; 9: 315-47.
- Khosroshahi A, Carruthers MN, Stone JH, et al. Rethinking Ormond’s disease idiopathic retroperitoneal fi brosis in the era of IgG4-related disease. Medicine Baltimore. 2013; 92: 82–91.
- Saeki T, Nishi S, Imai N, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010; 78: 1016–23.
- Takahashi N, Kawashima A, Fletcher J, et al. Renal involvement in patients with autoimmune pancreatitis: CT and MR imaging findings. Radiology. 2007; 242 (3):791-801.
- Sato Y, Takeuchi M, Takata K, et al. Clinicopathologic analysis of IgG4-related skin disease. Mod Path. 2012; 26: 523-32.
- Nishino T, Toki F, Oyama H, et al. Long term outcome of autoimmune pancreatitis after oral prednisolone therapy. Intern Med. 2006; 45: 497-501.
- Boonstra K, Culver EL, de Buy Wenniger LM, et al. Serum IgG4 and IgG1 for distinguishing IgG4-associated cholangitis from primary sclerosing cholangitis. Hepatology. 2014; 59: 1954–63.
- Wallace ZS, Mattoo H, Carruthers MN, et al. Plasmablasts as a biomarker for IgG4-related disease independent of serum IgG4 concentrations. Ann Rheum Dis. 2014 Jan; 74(1): 190-5.
- Kamisawa T, Shimosegawa T, Okazaki K, et al. Standard steroid treatment for autoimmune pancreatitis. Gut. 2009; 58: 1504-7.
- Khosroshahi A, Bloch DB, Deshpande V, et al. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4- related systemic disease. Arthritis Rheum. 2010; 62: 1755-62.