
Inherited LRP2 dysfunction in human disease and animal models
Renata Kozyraki1* and Olivier Cases1
1INSERM UMRS_1138, Centre de Recherche des Cordeliers, Paris-Diderot University, France
Abstract
Gp330/Megalin/Low-Density Lipoprotein Receptor-Related Protein 2 (LRP2) is an endocytic receptor that plays multiple roles in embryonic and adult tissues. It allows the cellular uptake of various bioactive molecules, morphogens, vitamins and hormones. Lack or dysfunction of the receptor affects renal protein reabsorption, lung function, brain and eye development in both man and experimental models. Mutations in LRP2 cause the polymalformative Donnai-Barrow syndrome, a rare autosomal recessive condition, combining developmental delay, facial dysmorphology, hearing defects, high myopia and low-molecular weight proteinuria.
We here summarize current knowledge on the receptor action. We particularly focus on the LRP2-associated face and eye anomalies and discuss how the receptor and its interacting proteins, including the multiligand receptor Cubilin (CUBN) may promote health or cause disease.
Introduction
Endocytosis is an essential mechanism that allows selective cellular uptake of numerous macromolecules. Receptor-mediated endocytosis provides the cell with nutrients and other bioactive molecules necessary for cellular metabolism and communication. During receptor-mediated endocytosis transmembrane cell surface receptors and their ligands enter endosomes from the plasma membrane. The ligands are either retained in the endosome and ultimately degraded in lysosomes or exported from the endosome and further reused. The balance between protein degradation and recycling is indispensable for cellular homeostasis and inappropriate sorting ultimately results in cellular and organ dysfunction.
LRP2, an endocytic receptor of the LDL receptor gene family
Gp330/Megalin was initially identified as the autoantigen in Heymann nephritis. The large plasma membrane receptor of 600 kDa was later characterized as a member of the low-density lipoprotein receptor (LDLR) gene family and called lipoprotein-related receptor protein 2 (LRP2). In man the gene is mapped to chromosome 2 (2q24-q31). It is around 235 kb long and mutations in LRP2 are at the origin of the autosomal recessive disorder Donnai-Barrow or facio-oculo-acoustico-renal syndrome (DBS/FOAR, see below)1,2.
LRP2 is an interspecies conserved, single-spanning membrane protein. The extracellular domain of the human protein consists of 4398 amino acids3. It is organized in four domains containing the so-called ligand binding repeats, cysteine-rich complement-type repeats shown to coordinate binding of the various LRP2 ligands and calcium. The intracellular domain is formed by 209 amino acids and contains 2 copies of the NPXY motif involved in receptor endocytosis and sorting. The interaction between NPXY-containing regions and adaptor proteins such as clathrin, AP2 adaptor complex, Autosomal Recessive Hypercholesterolemia protein (Low Density Lipoprotein Receptor Adapter Protein 1), Disabled-2 or the myosin VI motor protein4,5 is necessary for coated pit formation, ligand endocytosis and intracellular trafficking.
Expression pattern of LRP2
LRP2 is strongly expressed during development and in the adult. Both in man and mouse, LRP2 is detected at the maternal-fetal interfaces of the trophectoderm or the placenta as well as in numerous epithelia including the neuroepithelium, lung, thyroid, ocular and renal epithelia. In the adult the epithelial expression of LRP2 is globally preserved and is particularly strong in the gut and the absorptive epithelia of the renal proximal convoluted tubule (PCT), the reproductive organs, the ocular epithelia and the choroid plexus. Whereas LRP2 is exclusively detected at the apical plasma membrane of the PCT, pulmonary, intestinal or choroid plexus cells, in some cases, including the retinal pigment epithelium (RPE) cells, LRP2 is also expressed at the basolateral pole exposed to the systemic circulation. At the ultrastructural level the signal is distributed along the apical and/or basolateral endocytic vesicles, the recycling compartment and to some degree in lysosomes.
Functional interaction of LRP2 and Cubilin
In the above epithelia LRP2 functions together with Cubilin (CUBN), a structurally unrelated multiligand receptor that lacks a transmembrane domain (Figure 1)6,7. CUBN was previously identified as the physiological vitamin B12 receptor and mutations in CUBN result in a rare pediatric condition, the Imerslund-Grasbeck syndrome characterized by megaloblastic anemia, neurological disorders and proteinuria8.
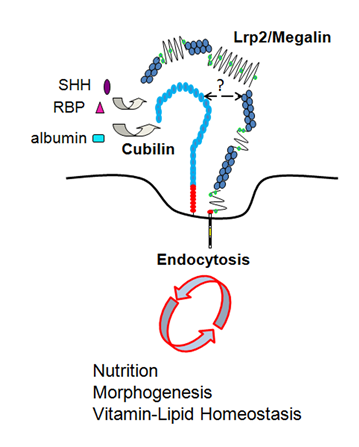
Figure 1.Apical endocytosis of nutrients and morphogens via Lrp2/megalin and Cubilin receptors. The exact interacting domains between Lrp2 and Cubn are not known but Cubn oligomers are thought to interact with one or more Lrp2 molecules.
The endocytic function of these proteins (LRP2-mediated, CUBN-assisted) has particularly been studied in the PCT where LRP2 and CUBN are the key receptors for the reabsorption and clearance of plasma proteins from the glomerular filtrate (Figure 2A)9. Among them are plasma protein carriers such as albumin, transthyretin, hemoglobin, leptin, angiotensin II, insulin, transferrin, various apolipoproteins, or hormones. Plasma vitamin carriers include the vitamin D binding protein (DBP), the retinol binding protein (RBP) or the vitamin B12 transporter transcobalamin (TCII). Retrieval of the carrier-vitamin complexes prevents urinary loss and conserves plasma levels of these essential metabolites (Figure 2A).
More than preventing vitamin D loss, LRP2 and CUBN are critical for its renal activation, a process that involves two hydroxylation steps. The first step occurs in the liver where vitamin D is hydroxylated to 25-hydroxyvitamin D3 (25(OH)-D3). The second one, i.e. transformation of 25(OH)-D3 to 1,25(OH)-D3, involves LRP2/CUBN-mediated renal uptake of circulating 25(OH)-D3 bound to DBP10,11. It is possible that LRP2 and CUBN expressed in the prostate, thyroid or breast cells may have a similar function in local vitamin D activation. CUBN physically interacts with LRP2 and DBP=25(OH)-D3 complexes enhancing thus their endocytosis; a similar mechanism may be applied to other common LRP2 and CUBN ligands.
Additionally, internalization of CUBN-specific ligands such as transferrin or high-density lipoproteins seems to be also LRP2-dependent. Urinary loss of CUBN and its ligands are constantly observed in the absence or dysfunction of LRP2 in man and animal models2,12–14. In the lung LRP2 and CUBN are expressed by the type II alveolar cells. Clearance of at least albumin, insulin or apolipoproteins at the alveolo-capillary barrier may thus also be LRP2/CUBN mediated and its impairment could play a role in acute lung injury and its more severe form the acute respiratory distress syndrome15.
Distinct fates of LRP2 ligands
The function of LRP2 as a clearance receptor for multiple ligands is established (Figure 2). Nevertheless, the fate of the internalized ligands may vary depending on the cellular context and the systemic needs. In most cases ligand internalization occurs through clathrin-coated pits at the apical plasma membrane; the ligand (i.e. DBP, RBP, TCII) is generally directed to the endosomal/lysosomal compartment for catabolism whereas the receptor is recycled back to the plasma membrane. Around 10% of the apically internalized ligand remains bound to LRP2 and moves together with it through the recycling compartment back to the apical membrane where it is re-secreted (proposed for the signaling molecule Sonic hedgehog SHH)16. Finally, the apical to basolateral plasma membrane transcytosis route was also described for some ligands including thyroglobulin and albumin17,18.
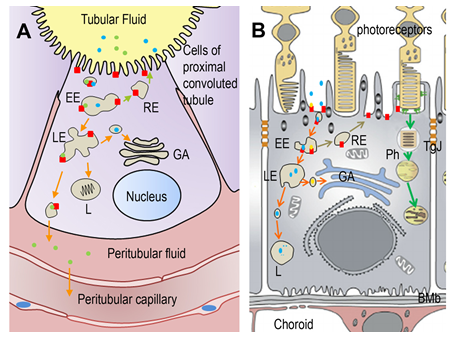
Figure 2. (A) Low-molecular weight proteins (albumin, transferrin) and polypeptides (hormones) present in the tubular fluid are taken up by an extensive network of coated pits in the proximal convoluted tubule. Two broad-spectrum binding proteins, Lrp2 and cubilin forming a complex (red square) recognize and bind filtered proteins and direct them to the coated pits. Then, molecules are taken up into vesicles by receptor-mediated endocytosis. Complexes accumulate in early endosomes (EE), the first cellular sorting station, where the cargo is distributed to the late endosomes (LE) or back to the plasma membrane (recycling endosome RE). Complexes which are destined for degradation in lysosomes (L) are transported from EE to L via LE. At LE vesicles can still enter the recycling/secretory pathway via Golgi apparatus (GA). A third route involves the complex trafficking to the baso-lateral membrane, resulting in the transcytosis of the ligand. Ligands present in the peritubular fluid can be taken up by the general circulation. (B) Albumin and transferrin are the main proteins found in the subretinal fluid. Lrp2-Cubilin complexes are expressed at the apical domain of retinal pigmented epithelial (RPE) cells. Beside phagocytosis of outer segments of the photoreceptors where Lrp2-Cubn may have a role, vesicles-coated endocytosis are observed at the basis of the apical microvilli. Vesicles are first transported into EE where cargo destined for degradation is sorted to their destination. From there, endosomes progress to LE and L for degradation or recycling back (RE) to the plasma membrane. Transcytosis to the choroidal circulation involving Lrp2-Cubn has not been characterized in RPE cells. BMb: Bruch’s membrane; GA: Golgi apparatus; Ph: phagosome; TgJ: tight junction.
LRP2-mediated albumin transcytosis in astrocytes is thought to be a key step for brain development by stimulating the synthesis and release of the neurotrophic factor oleic acid18. In this particular case however, LRP2-albumin internalization does not follow the classical clathrin-mediated route but appears to be caveolae-dependent.
Genetic LRP2 dysfunction in human pathology
The spectrum of Donnai Barrow syndrome
Whereas LRP2 dysfunction has been suggested in several acquired and common disorders such as chronic kidney disease, diabetes or hypertension the inherited dysfunction of LRP2 leads to a restricted number of pathologic conditions, namely the rare DBS. Among the 27 cases previously reviewed in19 (and references therein), 10 were diagnosed following termination of pregnancy for fetal anomaly or perinatal death secondary to other complications. More recently 17 novel cases were identified20–29. Most of the surviving patients present with a developmental delay but have no motor difficulties. All of them are characterized by a distinct set of craniofacial findings including hypertelorism, large anterior fontanelle, enlarged eye globes and pathological myopia, sensorineural hearing loss and complete or partial agenesis of the corpus callosum. The intellectual disabilities observed are mild to moderate but formal assessment is hampered by the visual and hearing defects. Bilateral, high-grade myopia (up to -23 diopters), retinal dystrophy, normal intraocular pressure, and occasionally iris coloboma and vitreous opacities are the ophthalmological findings in DBS patients. Progressive vision loss due to high myopia (HM) and the ensuing retinal detachment is common in the affected individuals. Prophylactic peripheral barrier photocoagulation has been helpful to prevent retinal detachment and improves chances of useful vision30. Regular monitoring of the hearing function is also necessary and hearing aids including cochlear implants are proposed to the patients20,21.
Low molecular weight proteinuria is a constant finding. Increased urinary excretion of DBP and RBP is pathognomonic and may be accompanied by lower serum levels of vitamin A and D. Diaphragmatic hernia and/or omphalocele are observed in 50% of the cases and are surgically treated at birth. Literature reports of some cases with heart anomalies such as persistence of the left canal vein or ventricular septal defects. Rare genitourinary system problems, focal segmental glomerulosclerosis and nephrotic range proteinuria are also occasionally described19. Self-injurious behavior has once been reported, a trouble most probably directly associated with the disease rather than being secondary to the developmental delay25. Brain anomalies such as periventricular nodular heterotopia or abnormalities of gyral patterns may occur and patients are often treated for seizures2,26.
Several disease-causing mutations are identified; they include missense, splice-site or nonsense mutations found throughout the Lrp2 gene but no phenotype-genotype correlations are currently available. Most mutations are predicted to result in protein loss or in a protein that abnormally accumulates in the intracellular space26. Renal biopsy of DBS patients does not show major morphological anomalies but reveals the presence of minor glomerular changes. The increased urinary loss of RBP, DBP but also CUBN, albumin and transferrin are associated with a defective formation of the apical endocytic machinery and a reduced number of endocytic and recycling vesicles. Consistent with the above observations classical proteinuria treatments including angiotensin-converting enzyme inhibitors are overall ineffective. Current data suggest that despite a urinary loss the renal function of DBS patients may remain normal. However long-term data on the evolution of the disease are missing and the oldest patient reported was 21-year-old. A progressive decline of the estimated glomerular filtration rate associated to the dysfunction of LRP2 cannot be excluded. Experimental evidence does not support a critical function of LRP2 during nephrogenesis31. Nevertheless, it is conceivable that minor modifications in the number of nephrons during embryonic development or altered endocytic activity of the LRP2-expressing podocytes, necessary for the formation of the glomerular filtration barrier, may facilitate the decline of the renal function and possibly result in renal insufficiency in the adult.
LRP2 dysfunction and its impact to other human disorders
A relationship of LRP2 with the Stickler syndrome, a rare connective tissue disorder, was recently reported in two Iraqi patients27. The above patients had several DBS features including proteinuria and increased urinary loss of beta2-microglobulin, a marker of tubular dysfunction and an established LRP2 ligand. The urinary excretion of RBP and DBP was however not evaluated in this study. It remains thus unclear whether the pathology described in these patients was the DBS or a Stickler-like syndrome.
Other pathologies that may result from LRP2 absence or dysfunction are autism spectrum disorders (ASD)32 and non-syndromic autosomal recessive intellectual disability33 characterized by defective memory and difficulties in language development. LRP2 is also believed to be a candidate locus for holoprosencephaly, i.e. incomplete cleavage of the prosencephalon. Haploinsufficiency of LRP2 can indeed contribute to holoprosencephaly microforms with mild facial features but no discernible brain anomalies34. Recently a single polymorphisme in LRP2 was reported in neural tube defects (NTD), congenital malformations due to defective neural tube closure. Interestingly, multiple CUBN polymorphisms were also associated with modified NTD risk35,36. More recently, congenital heart disease was also associated with a de novo mutation in LRP237 and Lrp2 was proposed to be a cilium-related gene38.
Finally, impaired LRP2 function has also been reported in Dent’s disease (MIM no. 300009) and in Lowe (oculo-cerebro-renal) syndrome (MIM no.309000) pathological conditions that affect the endosomal/lysosomal compartments, modify the expression of LRP2 and CUBN and lead to low-molecular weight proteinuria and, in the Lowe syndrome, to vision defects. The numerous ligand binding capacities and the multiple sites of LRP2 expression are compatible with the broad spectrum of phenotypes that accompany its lack or dysfunction. It remains unclear whether they are all directly associated with LRP2 but some clues are provided through the study of animal models with genetic disruption of the Lrp2 gene.
Animal models of Lrp2 dysfunction
Several animal models carrying targeted germ-line, N-ethyl-N-nitrosurea (ENU) induced or conditional disruption of the Lrp2 gene in mice or the zebrafish have been reported (Table1).
Table 1: Lrp2 inactivation in mouse and zebrafish models.
| Mouses | |||||
|---|---|---|---|---|---|
| Line | Molecular status | Target | Phenotype | Reference | |
| Lrp20/0 | Knock-out | Embryonic and extraembryonic tissues | Congenital diaphragmatic hernia, omphalocele, facial dysmorphy, abnormal oligodendroglial development, ACC, neural tube defect, cardiovascular abnormalities, impaired development of reproductive organs, LMWP | 31,39,41–43,45,52,59 | |
| Lrp2-L267 | Enu: stop codon to 2721 residue | Hypertrophy of choroid plexuses, enlarged ventricular system, loss of selenoprotein uptake in brain and kidney | 46,48,49 | ||
| Lrp2-L27 | Enu: missense mutation Cys4032Ser | Lack of forebrain dorsal commissures, hypertrophy of choroid plexuses | 47 | ||
|
Meox2-cre Lrp2lox/lox |
Conditional | Embryonic tissue | Cranial and telencephalic abnormalities, LMWP | 40,44,54 | |
|
FoxG1 : cre Lrp2lox/lox |
Conditional | Anterior forebrain | High myopia, retinal thinning, ACC | 57,60 | |
|
ApoE-cre Lrp2lox/lox |
Conditional | Kidney | Hypocalcemia, osteomalacia, LMWP | 68,69 | |
| Wnt4-Cre Lrp2lox/lox | Conditional | Kidney | LMWP | 70 | |
| Tie2-Cre Lrp2lox/lox | Conditional | Brain endothelium | Obesity, neurodegeneration, neuroinflammation | 71,72 | |
| Zebrafish | |||||
|
lrp2mw1 lrp2p5bnc |
Knock-out | Embryonic tissues | High myopia, glaucoma, renal reabsorption defects | 55,56 | |
| Morphant | Knock-down | Embryo | Renal reabsorption defects | 73 | |
ACC: agenesis of the corpus callosum, LMWP: low molecular weight proteinuria.
Lrp2 null mutants display several developmental and functional defects
Lrp2 germ-line inactivation results in defective forebrain and facial development. Holoprosencephaly, enlarged lateral ventricles, cortical thinning or exencephaly, a cranial neural tube defect, are frequently observed in the mutants. The facial anomalies include extreme microphthalmia/anophthalmia, flattened forehead and shortened nose and are suggestive of impaired cephalic neural crest functions. Perturbed spinal cord development including of glial cells, reproductive organ and limb anomalies, impaired pulmonary, cardiac and kidney function complete the mutant phenotype. Cardiovascular defects include aortic arch anomalies, persistent truncus arteriosus and ventricular anomalies, findings that corroborate the cardiac phenotype observed in DBS patients. The alveolar defects cause respiratory insufficiency and explain the increased perinatal lethality of the mutants with only around 2-5% of mice surviving to adult age31,39–45.
ENU-induced disruption of Lrp2 partially mimics the Lrp2 null phenotype
A single basepair substitution in Lrp2 most likely resulting in a soluble, truncated Lrp2 consisting of the aminoterminal portion of the extracellular domain was first reported46. In a second study, the ENU treatment led to a protein carrying a Cys4032Ser missence mutation probably affecting the ligand binding capacities of LRP247. These mutants present craniofacial defects including anophthalmia, an enlarged ventricular system, abnormalities in the dorsal telencephalon, defective neurogenesis, impaired movement coordination, axon guidance defects and proteinuria46,48,49. HPE was not a constant finding in the above cases.
A third ENU mutagenesis approach recently recognized Lrp2 as a cilium-related gene associated with congenital heart disease38. In this mutant a c.8456-3A>G nucleotide change affects the extra-cellular domain of Lrp2 and possibly results in a truncated protein. Although the available immunomorphological data do not support a ciliary function of Lrp245, the implication of Lrp2 in cardiovascular development is consistent with human and animal studies.
There is a phenotypic discrepancy between the ENU- and Lrp2 null mutants. It is possible that the defective proteins may retain some residual functions. Alternatively and as proposed by Zarbalis et al46, in the Lrp2 null mice additional proteins may also be affected, a hypothesis consistent with the presence of an LRP2 receptor network.
Nutritional and signaling defects associated with Lrp2 dysfunction in the neuroectoderm and neural-crest derived structures
Defective cholesterol and/or vitamin D and/or vitamin A uptake in the LRP2-deficient neuroectoderm may partly explain the forebrain and facial defects of the mutants. Additionally, LRP2 may regulate craniofacial development through its interactions with the SHH signaling pathway. During early development, forebrain ventral midline structures are specified by SHH signals originating from the prechordal plate and the ventral forebrain neuroectoderm (ventral patterning center). These signals induce the formation of ventral cell populations and antagonize bone morphogentic protein (BMP) signaling from the dorsal patterning center. Experimental evidence indicates that LRP2 expressed in the neuroepithelium overlying the prechordal plate forms a co-receptor complex with Patched homolog-1 (PTCH1), the canonical SHH receptor, facilitating its endocytosis, relieving PTCH1 dependent suppression of Smoothend and thus activating the SHH pathway44. Lack of LRP2 may affect SHH-dependent cell proliferation in the ventral forebrain but may also affect BMP signaling by acting as a clearance receptor for BMP440. At later stages a disturbed SHH-BMP balance may also explain the defective astrocytic differentiation or the impaired neurogenesis observed in the Lrp2 mutants41,49.
Another molecule essential in dorso-ventral patterning of the rostral head and facial morphogenesis is fibroblast growth factor (FGF8) secreted from the rostral patterning center. FGF8 secretion promotes among others migration, survival and patterning of the cephalic neural crest cells, a population indispensable for neural tube closure, normal cephalic vesicle formation and facial morphogenesis. FGF8 is a high affinity ligand for CUBN and CUBN-dependent FGF8 endocytosis promotes FGF signaling through the phosphorylation and activation of the ERK pathway50. The underlying mechanism of ERK1/2 phosphorylation remains unclear but CUBN may sequestrate FGF8 in the endosomal compartment and thus facilitate its interaction with its cognate FGF receptors. CUBN and LRP2 have overlapping expression patterns in the cephalic neural crest cells as well as in the rostral neuroepithelium, tissues that do not express the other CUBN partner Amnionless (reviewed in7). It can be assumed that in the above sites, as in the kidney, CUBN internalization may partly or fully depend on LRP2. In this case LRP2 deficiency would impair CUBN function by limiting or even abolishing the endocytosis of CUBN-FGF8 complexes, indirectly interfering with FGF8 signaling. This observation is compatible with the impaired FGF8 expression in the LRP2 mutants and may contribute to the previously described loss of SHH in the preoptic area13,14,50,51. Experimental evidence obtained in the mouse, chick and Xenopus models indicates that CUBN is indeed required for anterior head cell survival and morphogenesis by modulating the equilibrium between the antagonistic pro- and anti-apoptotic BMP and FGF activities50. It is therefore tempting to propose that in the Lrp2 null mice deficient CUBN function, including in cholesterol and vitamin D uptake, contributes to the observed forebrain and facial abnormalities.
Similar mechanisms may at least partly explain the impaired development of other neural crest-derived structures including the cardiovascular defects observed in Lrp2 mutants45. Indeed, impaired SHH or FGF8 signaling may both explain the persistent truncus arteriosus and the ventricular septal defects. Alternatively, LRP2 dysfunction may disturb RBP-vitamin A cellular uptake and consequently retinoic acid signaling. Supporting this hypothesis the cardiac phenotype of patients with mutations in the STRA6 gene (the main RBP receptor in humans) resembles to the one of DBS patients.
Lrp2 in steroid hormone metabolism
LRP2 is expressed in the adult male and female reproductive organs and is involved in the cellular uptake of biologically active androgens and estrogens bound to their carrier the sex hormone binding globulin (SHBG)51,52. Following endocytosis SHBG is degraded and the released steroids can induce their target genes. Despite normal systemic androgen and estrogen levels, testicular maldescent and vaginal obstruction are observed in the Lrp2 null mice supporting the idea that the LRP2-mediated route is critical for sexual maturation52. The exact molecular mechanism remains unclear but it is possible that LRP2 partially facilitates the activity of steroid hormones, namely the induction of the apoptotic processes involved in opening the vagina cavity and the testicular descent. LRP2 is also expressed in epithelial cells of the mammary gland and the prostate and may thus contribute to sex steroid uptake by these organs under normal and pathological conditions.
Tissue-specific inactivation of Lrp2 in mice
Meox2::Cre mediated mosaic inactivation of Lrp2 in the epiblast leads to a an Lrp2 null-like phenotype clearly indicating the necessity of LRP2 in the embryonic but not the extra-embryonic (i.e. trophectoderm, visceral yolk sac, placenta) tissues40.
Kidney-specific inactivation of the Lrp2 gene leads to proteinuria
Several studies have described the function of LRP2 in the kidney (Table1) and have recently been reviewed53. Despite the fact that the inactivation of Lrp2 or Cubn in this tissue was never complete the loss of these proteins resulted in deficient proximal tubular reabsorption, impairment of the apical endocytic apparatus and proteinuria. Several of the excreted proteins are specific LRP2 and/or Cubn ligands including DBP, RBP, TCII or albumin. The tubular reabsorption of these and other essential metabolites contributes to systemic vitamin, hormone and lipid homeostasis39,54.
Deficient renal reabsorption is one of the major phenotypes of the zebrafish model organism with lack or dysfunction of the lrp2 gene55 (Table1) indicating an evolutionary over species conserved endocytic function of the receptor. It is of interest that no brain anomalies are found in this model. However, in the naturally occurring zebrafish lrp2 mutant, bugeye, adult-onset eye enlargement is observed. The origin of this feature is not clear but may be due to elevated intra-ocular pressure56.
Conditional Lrp2 disruption in the anterior neuroepithelium and ocular tissues results in high myopia
Targeted FoxG1::Cre mediated Lrp2 ablation from the anterior neuroepithelium leads to absent corpus callosum and most interestingly in early onset excessive eye growth and HM reminiscent of the human pathology. In the homozygous mutant mice increased axial length (the endophenotype of myopia) chorioretinal atrophy and posterior staphyloma together with a decreased number of retinal ganglion and photoreceptor cells are observed57.
Bilateral or unilateral microphthalmia or anophthalmia are commonly seen in germ-line inactivated Lrp2 mutants. In contrast, in FoxG1::Lrp2 mutants where Lrp2 inactivation occurs after the eye field and optic vesicle are formed around embryonic day 9, excessive bilateral eye enlargement is the principal defect57. The origin of the above diametrically opposed phenotypes is elusive. During eye development LRP2-mediated endocytosis and degradation of SHH was proposed to be necessary for the normal formation and function of the retinal and ciliary margin cells58. Loss of this regulatory step in the absence of LRP2 was consequently thought to result in eye enlargement. However, retinal and ciliary body development are preserved in the Foxg1::Lrp2 mutants an observation compatible with normal expression of LRP2 until mid-gestation57. Additionally, eye enlargement and retinal thinning are not observed before the first week of post-natal life arguing against a central role of the LRP2-SHH interaction in the ciliary margin, in myopia induction. Nevertheless, impaired LRP2-mediated SHH transcytosis and signaling in the FoxG1::Lrp2 mutants may result in defective migration of the oligodendrocyte precursor cells from the preoptic area to the optic nerve early after birth and explain the reduced number of oligodendrocytes in the optic nerves of the mutants57,59.
The ionic equilibrium is modified in the vitreous of Lrp2-deficient eyes
Further analysis of the LRP2-deficient myopic mouse eyes reveals that the increased axial length is almost exclusively due to the enlargement of the mutant vitreal chamber60. Under normal conditions in both man and mice the vitreous chamber is mainly filled with the vitreous humor, a highly hydrated gel like structure essential for normal refractive growth. In highly myopic eyes the vitreous gel volume is progressively replaced by a liquid vitreous, a process known as vitreous liquefaction61. Vitreous liquefaction is closely associated with the progression of the HM-associated pathologies nuclear cataract, glaucoma and rhegmatogenous retinal detachment62. Moreover, experimental evidence obtained in the chick suggests that vitreous liquefaction may also be at the origin of the excessive eye growth63.
In the LRP2-deficient eyes the vitreal chamber contains a normally formed vitreous gel and in addition an extremely high amount of an acellular liquid fraction (LF). The ionic composition of the LF shows major modifications in the mutants: the abundance of potassium, sodium and chloride are significantly decreased and the concentration of phosphorus and calcium are abnormally increased. These modifications most likely reflect impaired electrolytic equilibrium in the surrounding retina and RPE and may at least partly explain the aberrant ocular growth64. Deficient, ionically driven trans-retinal liquid flow between the vitreous chamber and the choroidal vasculature may physically result in fluid accumulation in the vitreous chamber. RPE and Müller cells, the two cell types that normally express LRP2, are central in this process and reduction of fluid outflow across the RPE, secondary to the LRP2 absence and the possibly ensuing RPE/Müller cell dysfunction could lead to the increase of the vitreous volume. It is possible that in these cell types, as in the kidney, LRP2 controls the sodium and phosphate homeostasis via its interactions with the Na+/H+ exchanger or the type IIa sodium phosphate co-transporter65. Whether, however, the lack of LRP2 directly or indirectly influences fluid movement across the RPE and retinal cells has not yet been investigated.
Modified vitreal proteome and defective endocytosis in the Lrp2-deficient eyes
In addition to the ionic disequilibrium, the proteome of the LF is also dramatically modified in the mutant eyes. A limited number of proteins, mainly established LRP2 and CUBN ligands such as albumin, DBP, RBP, transferrin, apolipoprotein A-I, TCII, transthyretin or carbonic anhydrase 1 are over-expressed in the LRP2-deficient eyes. It is of interest that abnormally high levels of several of the above proteins, including the myopia-associated signal apolipoptrotein A-I, are found in the ocular fluids of patients with HM66. The above proteins either derive from the systemic circulation or are locally synthesized including by the RPE and ciliary epithelium. It is therefore reasonable to assume that impaired handling of the above proteins in the ocular epithelia due to the absence or dysfunction of LRP2-mediated endocytosis may be a cause of eye enlargement. Consistent with this hypothesis, the decreased or even absent expression of the early endocytic and lysosomal markers EEA1, cathepsin D and LIMP2 (Lysosome membrane protein 2), essential for lysosome biogenesis, indicates that endosomal and lysosomal formation are compromised in the LRP2-deficient RPE60. In addition to the inappropriate protein degradation, the disturbances in the lysosomal pathway and the quasi loss of LIMP2 directly affect the transport of potassium67 and could therefore also contribute to the impaired ionic composition of the mutant LF. Furthermore, the basolateral uptake of intravenously injected albumin or transferrin (a specific CUBN ligand) is abolished in the LRP2-deficient RPE clearly confirming that LRP2-, and presumably CUBN-, mediated clearance of serum-derived proteins is an essential function of the RPE cells. How this (dys)function may be associated with myopia induction and/or progression is currently under investigation. Finally, LRP2 seems to have additional functions at the apical pole of the RPE cells: the absence of apical microvilli and the reduced formation of junctional complexes support the idea that LRP2 is essential for the establishment or maintenance of the apical RPE polarity. The RPE facing outer rod segments of the mutant eyes are also absent. This anomaly may be related to the reduced exchange/clearance capacities of the mutant RPE and/or the complete downregulation of the main retinoic acid synthesizing enzyme RALDH2 in the mutant eyes. Independently on the cause of RALDH2 absence the ensuing impaired retinol homeostasis is likely to contribute in the aberrant post-natal eye growth.
It is likely that modifications in the SHH-, BMP- or FGF-dependent morphogenetic pathways during early pre-natal eye development may explain some of the ocular phenotypes associated with LRP2, i.e. microphthalmia/anophthalmia. At later developmental stages and in the adult LRP2-mediated plasma protein and vitamin endocytosis appears to regulate normal and pathological eye growth. Future work is necessary to establish the mechanism(s) of myopia induction in the LRP2 mutants and elucidate whether the observed retinal degenerations, scleral and choroidal modifications in both the DBS patients and the FoxG1::lrp2 mutants are directly or not the outcome of LRP2 absence.
Conclusion
Published work clearly shows that LRP2 has site- and time-specific functions. These functions are established in the adult kidney whereas its role as a morphogen receptor or a regulator of adult eye growth is emerging. The central role of LRP2 is the constitutive clearance of its ligands and CUBN may be an essential LRP2 partner both in the adult and the embryonic epithelia. Depending on ligand availability LRP2-mediated, CUBN-assisted endocytosis may promote health or disease including in the kidney either by reducing renal reuptake of nephrotoxic agents or by enhancing the uptake of protective factors.
Experimental evidence suggests that in the eye and other extra-renal tissues, synergistic LRP2 and CUBN activities may be required. However, in human pathology mutations in LRP2 cause the severe DBS syndrome whereas mutations in CUBN lead to a relatively mild condition, the Immerslund-Gräsbeck syndrome. On the other hand reduced CUBN or LRP2 function are associated with NTDs suggesting that the complex LRP2-CUBN interactions are not fully understood and that additional, yet to be identified, partners may be involved. Further studies are necessary to clearly establish the cellular and molecular interdependence of these receptors and clarify their potential as preventive or even therapeutic targets.
References
-
- Korenberg JR, Argraves KM, Chen XN, Tran H, Strickland DK, Argraves WS. Chromosomal localization of human genes for the LDL receptor family member glycoprotein 330 (LRP2) and its associated protein RAP (LRPAP1). Genomics. 1994;22(1):88-93. doi:10.1006/geno.1994.1348.
- Kantarci S, Al-Gazali L, Hill RS, et al. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat Genet. 2007;39(8):957-959. doi:10.1038/ng2063.
- Hjälm G, Murray E, Crumley G, et al. Cloning and sequencing of human gp330, a Ca(2+)-binding receptor with potential intracellular signaling properties. Eur J Biochem. 1996;239(1):132-137.
- Gallagher H, Oleinikov AV, Fenske C, Newman DJ. The adaptor disabled-2 binds to the third psi xNPxY sequence on the cytoplasmic tail of megalin. Biochimie. 2004;86(3):179-182. doi:10.1016/j.biochi.2004.03.001.
- Morris SM, Tallquist MD, Rock CO, Cooper JA. Dual roles for the Dab2 adaptor protein in embryonic development and kidney transport. EMBO J. 2002;21(7):1555-1564. doi:10.1093/emboj/21.7.1555.
- Moestrup SK, Kozyraki R, Kristiansen M, et al. The intrinsic factor-vitamin B12 receptor and target of teratogenic antibodies is a megalin-binding peripheral membrane protein with homology to developmental proteins. J Biol Chem. 1998;273(9):5235-5242.
- Kozyraki R, Cases O. Vitamin B12 absorption: mammalian physiology and acquired and inherited disorders. Biochimie. 2013;95(5):1002-1007. doi:10.1016/j.biochi.2012.11.004.
- Kozyraki R, Kristiansen M, Silahtaroglu A, et al. The human intrinsic factor-vitamin B12 receptor, cubilin: molecular characterization and chromosomal mapping of the gene to 10p within the autosomal recessive megaloblastic anemia (MGA1) region. Blood. 1998;91(10):3593-3600.
- Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol. 2002;3(4):256-266. doi:10.1038/nrm778.
- Nykjaer A, Dragun D, Walther D, et al. An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D3. Cell. 1999;96(4):507-515.
- Nykjaer A, Fyfe JC, Kozyraki R, et al. Cubilin dysfunction causes abnormal metabolism of the steroid hormone 25(OH) vitamin D(3). Proc Natl Acad Sci U S A. 2001;98(24):13895-13900. doi:10.1073/pnas.241516998.
- Storm T, Zeitz C, Cases O, et al. Detailed investigations of proximal tubular function in Imerslund-Gräsbeck syndrome. BMC Med Genet. 2013;14:111. doi:10.1186/1471-2350-14-111.
- Kozyraki R, Fyfe J, Kristiansen M, et al. The intrinsic factor-vitamin B12 receptor, cubilin, is a high-affinity apolipoprotein A-I receptor facilitating endocytosis of high-density lipoprotein. Nat Med. 1999;5(6):656-661. doi:10.1038/9504.
- Kozyraki R. Cubilin, a multifunctional epithelial receptor: an overview. J Mol Med Berl Ger. 2001;79(4):161-167.
- Buchäckert Y, Rummel S, Vohwinkel CU, et al. Megalin mediates transepithelial albumin clearance from the alveolar space of intact rabbit lungs. J Physiol. 2012;590(20):5167-5181. doi:10.1113/jphysiol.2012.233403.
- Morales CR, Zeng J, El Alfy M, et al. Epithelial trafficking of Sonic hedgehog by megalin. J Histochem Cytochem Off J Histochem Soc. 2006;54(10):1115-1127. doi:10.1369/jhc.5A6899.2006.
- Marinò M, Lisi S, Pinchera A, Chiovato L, McCluskey RT. Targeting of thyroglobulin to transcytosis following megalin-mediated endocytosis: evidence for a preferential pH-independent pathway. J Endocrinol Invest. 2003;26(3):222-229.
- Bento-Abreu A, Velasco A, Polo-Hernández E, et al. Albumin endocytosis via megalin in astrocytes is caveola- and Dab-1 dependent and is required for the synthesis of the neurotrophic factor oleic acid. J Neurochem. 2009;111(1):49-60. doi:10.1111/j.1471-4159.2009.06304.x.
- Pober BR, Longoni M, Noonan KM. A review of Donnai-Barrow and facio-oculo-acoustico-renal (DB/FOAR) syndrome: clinical features and differential diagnosis. Birt Defects Res A Clin Mol Teratol. 2009;85(1):76-81. doi:10.1002/bdra.20534.
- Bruce IA, Broomfield SJ, Henderson L, Green KMJ, Ramsden RT. Cochlear implantation in Donnai-Barrow syndrome. Cochlear Implants Int. 2011;12(1):60-63. doi:10.1179/146701010X486534.
- Broomfield SJ, Bruce IA, Henderson L, Ramsden RT, Green KMJ. Cochlear implantation in children with syndromic deafness. Int J Pediatr Otorhinolaryngol. 2013;77(8):1312-1316. doi:10.1016/j.ijporl.2013.05.022.
- Khalifa O, Al-Sahlawi Z, Imtiaz F, et al. Variable expression pattern in Donnai-Barrow syndrome: Report of two novel LRP2 mutations and review of the literature. Eur J Med Genet. 2015;58(5):293-299. doi:10.1016/j.ejmg.2014.12.008.
- Storm T, Tranebjærg L, Frykholm C, et al. Renal phenotypic investigations of megalin-deficient patients: novel insights into tubular proteinuria and albumin filtration. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc - Eur Ren Assoc. 2013;28(3):585-591. doi:10.1093/ndt/gfs462.
- Chinta S, Gupta A, Sachdeva V, Kekunnaya R. Persistent pupillary membrane, strabismus, and optic nerve hypoplasia in Donnai-Barrow syndrome. J AAPOS Off Publ Am Assoc Pediatr Ophthalmol Strabismus. 2011;15(6):604-605. doi:10.1016/j.jaapos.2011.07.016.
- Roane H, Bouxsein K, Fulton C. Assessment and Treatment of Self-Injurious Behavior Associated with Donnai-Barrow Syndrome. J Dev Phys Disabil. 2012;24(4):327-335. doi:10.1007/s10882-012-9272-6.
- Dachy A, Paquot F, Debray G, et al. In-depth phenotyping of a Donnai-Barrow patient helps clarify proximal tubule dysfunction. Pediatr Nephrol Berl Ger. 2015;30(6):1027-1031. doi:10.1007/s00467-014-3037-7.
- Schrauwen I, Sommen M, Claes C, et al. Broadening the phenotype of LRP2 mutations: a new mutation in LRP2 causes a predominantly ocular phenotype suggestive of Stickler syndrome. Clin Genet. 2014;86(3):282-286. doi:10.1111/cge.12265.
- Shaheen IS, Finlay E, Prescott K, Russell M, Longoni M, Joss S. Focal segmental glomerulosclerosis in a female patient with Donnai-Barrow syndrome. Clin Dysmorphol. 2010;19(1):35-37. doi:10.1097/MCD.0b013e328333c20a.
- de Ligt J, Willemsen MH, van Bon BWM, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367(20):1921-1929. doi:10.1056/NEJMoa1206524.
- Patel N, Hejkal T, Katz A, Margalit E. Ocular manifestations of Donnai-Barrow syndrome. J Child Neurol. 2007;22(4):462-464. doi:10.1177/0883073807301933.
- Willnow TE, Hilpert J, Armstrong SA, et al. Defective forebrain development in mice lacking gp330/megalin. Proc Natl Acad Sci U S A. 1996;93(16):8460-8464.
- Ionita-Laza I, Makarov V, ARRA Autism Sequencing Consortium, Buxbaum JD. Scan-statistic approach identifies clusters of rare disease variants in LRP2, a gene linked and associated with autism spectrum disorders, in three datasets. Am J Hum Genet. 2012;90(6):1002-1013. doi:10.1016/j.ajhg.2012.04.010.
- Vasli N, Ahmed I, Mittal K, et al. Identification of a homozygous missense mutation in LRP2 and a hemizygous missense mutation in TSPYL2 in a family with mild intellectual disability. Psychiatr Genet. 2016;26(2):66-73. doi:10.1097/YPG.0000000000000114.
- Rosenfeld JA, Ballif BC, Martin DM, et al. Clinical characterization of individuals with deletions of genes in holoprosencephaly pathways by aCGH refines the phenotypic spectrum of HPE. Hum Genet. 2010;127(4):421-440. doi:10.1007/s00439-009-0778-7.
- Franke B, Vermeulen SHHM, Steegers-Theunissen RPM, et al. An association study of 45 folate-related genes in spina bifida: Involvement of cubilin (CUBN) and tRNA aspartic acid methyltransferase 1 (TRDMT1). Birt Defects Res A Clin Mol Teratol. 2009;85(3):216-226. doi:10.1002/bdra.20556.
- Pangilinan F, Molloy AM, Mills JL, et al. Evaluation of common genetic variants in 82 candidate genes as risk factors for neural tube defects. BMC Med Genet. 2012;13:62. doi:10.1186/1471-2350-13-62.
- Zaidi S, Choi M, Wakimoto H, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498(7453):220-223. doi:10.1038/nature12141.
- Li Y, Klena NT, Gabriel GC, et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature. 2015;521(7553):520-524. doi:10.1038/nature14269.
- Leheste JR, Rolinski B, Vorum H, et al. Megalin knockout mice as an animal model of low molecular weight proteinuria. Am J Pathol. 1999;155(4):1361-1370. doi:10.1016/S0002-9440(10)65238-8.
- Spoelgen R, Hammes A, Anzenberger U, et al. LRP2/megalin is required for patterning of the ventral telencephalon. Dev Camb Engl. 2005;132(2):405-414. doi:10.1242/dev.01580.
- Wicher G, Aldskogius H. Megalin deficiency induces critical changes in mouse spinal cord development. Neuroreport. 2008;19(5):559-563. doi:10.1097/WNR.0b013e3282f94267.
- Kur E, Mecklenburg N, Cabrera RM, Willnow TE, Hammes A. LRP2 mediates folate uptake in the developing neural tube. J Cell Sci. 2014;127(Pt 10):2261-2268. doi:10.1242/jcs.140145.
- Sabatino JA, Stokes BA, Zohn IE. Prevention of neural tube defects in Lrp2 mutant mouse embryos by folic acid supplementation. Birth Defects Res. 2017;109(1):16-26. doi:10.1002/bdra.23589.
- Christ A, Christa A, Kur E, et al. LRP2 is an auxiliary SHH receptor required to condition the forebrain ventral midline for inductive signals. Dev Cell. 2012;22(2):268-278. doi:10.1016/j.devcel.2011.11.023.
- Baardman ME, Zwier MV, Wisse LJ, et al. Common arterial trunk and ventricular non-compaction in Lrp2 knockout mice indicate a crucial role of LRP2 in cardiac development. Dis Model Mech. 2016;9(4):413-425. doi:10.1242/dmm.022053.
- Zarbalis K, May SR, Shen Y, Ekker M, Rubenstein JLR, Peterson AS. A focused and efficient genetic screening strategy in the mouse: identification of mutations that disrupt cortical development. PLoS Biol. 2004;2(8):E219. doi:10.1371/journal.pbio.0020219.
- Ha S, Stottmann RW, Furley AJ, Beier DR. A forward genetic screen in mice identifies mutants with abnormal cortical patterning. Cereb Cortex N Y N 1991. 2015;25(1):167-179. doi:10.1093/cercor/bht209.
- Chiu-Ugalde J, Theilig F, Behrends T, et al. Mutation of megalin leads to urinary loss of selenoprotein P and selenium deficiency in serum, liver, kidneys and brain. Biochem J. 2010;431(1):103-111. doi:10.1042/BJ20100779.
- Gajera CR, Emich H, Lioubinski O, et al. LRP2 in ependymal cells regulates BMP signaling in the adult neurogenic niche. J Cell Sci. 2010;123(Pt 11):1922-1930. doi:10.1242/jcs.065912.
- Cases O, Perea-Gomez A, Aguiar DP, et al. Cubilin, a high affinity receptor for fibroblast growth factor 8, is required for cell survival in the developing vertebrate head. J Biol Chem. 2013;288(23):16655-16670. doi:10.1074/jbc.M113.451070.
- Assémat E, Châtelet F, Chandellier J, et al. Overlapping expression patterns of the multiligand endocytic receptors cubilin and megalin in the CNS, sensory organs and developing epithelia of the rodent embryo. Gene Expr Patterns GEP. 2005;6(1):69-78. doi:10.1016/j.modgep.2005.04.014.
- Hammes A, Andreassen TK, Spoelgen R, et al. Role of endocytosis in cellular uptake of sex steroids. Cell. 2005;122(5):751-762. doi:10.1016/j.cell.2005.06.032.
- Eshbach ML, Weisz OA. Receptor-Mediated Endocytosis in the Proximal Tubule. Annu Rev Physiol. 2017;79:425-448. doi:10.1146/annurev-physiol-022516-034234.
- Amsellem S, Gburek J, Hamard G, et al. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J Am Soc Nephrol JASN. 2010;21(11):1859-1867. doi:10.1681/ASN.2010050492.
- Kur E, Christa A, Veth KN, et al. Loss of Lrp2 in zebrafish disrupts pronephric tubular clearance but not forebrain development. Dev Dyn Off Publ Am Assoc Anat. 2011;240(6):1567-1577. doi:10.1002/dvdy.22624.
- Veth KN, Willer JR, Collery RF, et al. Mutations in zebrafish lrp2 result in adult-onset ocular pathogenesis that models myopia and other risk factors for glaucoma. PLoS Genet. 2011;7(2):e1001310. doi:10.1371/journal.pgen.1001310.
- Cases O, Joseph A, Obry A, et al. Foxg1-Cre Mediated Lrp2 Inactivation in the Developing Mouse Neural Retina, Ciliary and Retinal Pigment Epithelia Models Congenital High Myopia. PloS One. 2015;10(6):e0129518. doi:10.1371/journal.pone.0129518.
- Christ A, Herzog K, Willnow TE. LRP2, an auxiliary receptor that controls sonic hedgehog signaling in development and disease. Dev Dyn Off Publ Am Assoc Anat. 2016;245(5):569-579. doi:10.1002/dvdy.24394.
- Ortega MC, Cases O, Merchán P, Kozyraki R, Clemente D, de Castro F. Megalin mediates the influence of sonic hedgehog on oligodendrocyte precursor cell migration and proliferation during development. Glia. 2012;60(6):851-866. doi:10.1002/glia.22316.
- Cases O, Obry A, Ben-Yacoub S, et al. Impaired vitreous composition and retinal pigment epithelium function in the FoxG1::LRP2 myopic mice. Biochim Biophys Acta. March 2017. doi:10.1016/j.bbadis.2017.03.022.
- Le Goff MM, Bishop PN. Adult vitreous structure and postnatal changes. Eye Lond Engl. 2008;22(10):1214-1222. doi:10.1038/eye.2008.21.
- Morgan IG, Ohno-Matsui K, Saw S-M. Myopia. Lancet Lond Engl. 2012;379(9827):1739-1748. doi:10.1016/S0140-6736(12)60272-4.
- Halfter W. Change in embryonic eye size and retinal cell proliferation following intravitreal injection of glycosaminoglycans. Invest Ophthalmol Vis Sci. 2008;49(8):3289-3298. doi:10.1167/iovs.07-1421.
- Crewther SG, Murphy MJ, Crewther DP. Potassium channel and NKCC cotransporter involvement in ocular refractive control mechanisms. PloS One. 2008;3(7):e2839. doi:10.1371/journal.pone.0002839.
- Bachmann S, Schlichting U, Geist B, et al. Kidney-specific inactivation of the megalin gene impairs trafficking of renal inorganic sodium phosphate cotransporter (NaPi-IIa). J Am Soc Nephrol JASN. 2004;15(4):892-900.
- Duan X, Lu Q, Xue P, et al. Proteomic analysis of aqueous humor from patients with myopia. Mol Vis. 2008;14:370-377.
- Knipper M, Claussen C, Rüttiger L, et al. Deafness in LIMP2-deficient mice due to early loss of the potassium channel KCNQ1/KCNE1 in marginal cells of the stria vascularis. J Physiol. 2006;576(Pt 1):73-86. doi:10.1113/jphysiol.2006.116889.
- Leheste JR, Melsen F, Wellner M, et al. Hypocalcemia and osteopathy in mice with kidney-specific megalin gene defect. FASEB J Off Publ Fed Am Soc Exp Biol. 2003;17(2):247-249. doi:10.1096/fj.02-0578fje.
- Motoyoshi Y, Matsusaka T, Saito A, et al. Megalin contributes to the early injury of proximal tubule cells during nonselective proteinuria. Kidney Int. 2008;74(10):1262-1269. doi:10.1038/ki.2008.405.
- Weyer K, Storm T, Shan J, et al. Mouse model of proximal tubule endocytic dysfunction. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc - Eur Ren Assoc. 2011;26(11):3446-3451. doi:10.1093/ndt/gfr525.
- Bartolome F, Antequera D, Tavares E, et al. Obesity and neuroinflammatory phenotype in mice lacking endothelial megalin. J Neuroinflammation. 2017;14(1):26. doi:10.1186/s12974-017-0800-2.
- Dietrich M, Antequera D, Pascual C, Castro N, Bolos M, Carro E. Alzheimer’s disease-like impaired cognition in endothelial-specific megalin-null mice. J Alzheimers Dis JAD. 2014;39(4):711-717. doi:10.3233/JAD-131604.
- Anzenberger U, Bit-Avragim N, Rohr S, et al. Elucidation of megalin/LRP2-dependent endocytic transport processes in the larval zebrafish pronephros. J Cell Sci. 2006;119(Pt 10):2127-2137. doi:10.1242/jcs.02954.