
Lenz-Majewski syndrome: How a single mutation leads to complex changes in lipid metabolism
Mira Sohn and Tamas Balla*
Section on Molecular Signal Transduction, Program for Developmental Neuroscience, Eunice Kennedy Shriver NICHD, National Institutes of Health, Bethesda, Maryland 20892, USA
Abstract
Lenz-Majewski syndrome (LMS) is a rare disease presenting with complex physical and mental abnormalities. Whole exome sequencing performed on five LMS-affected individuals has identified gain-of-function mutations in the PTDSS1 gene encoding phosphatidylserine synthase 1 (PSS1) enzyme. These mutations all rendered PSS1 insensitive to PS-mediated product inhibition. In a recent study we showed that uncontrolled PS production by these mutant PSS1 enzymes lead to the accumulation of PS in the ER where it is not detected in normal cells. This increased PS in the ER in turn, activated the Sac1 phosphatase, which is responsible for the dephosphorylation of the minor lipid, phosphatidylinositol 4-phosphate (PI4P) in the ER. Increased Sac1 activity decreased PI4P levels both in the Golgi and the plasma membrane thereby dissipating the PI4P gradients set up by PI 4-kinase enzymes (PI4Ks) between these membranes and the ER. Such PI4P gradients at membrane contact sites have been shown to support the transports of structural lipids such as cholesterol and PS out of the ER by non-vesicular lipid transfer. Therefore, uncontrolled production of PS not only affects the PS status of cells but also initiates an avalanche of changes in the metabolism of other membrane lipids via affecting PI4P gradients throughout the cell. Recognition of the close metabolic interaction between PS synthesis and PI4P metabolism provided a new clue to better understand the molecular underpinning of this rare and severe disease.
Lenz-Majewski Syndrome (LMS), also known as Lenz-Majewski hyperostotic dwarfism (LMHD), has been described originally by Lenz and Majewski in 1974 as a case report of a two-year-old child who presented with multiple developmental abnormalities including intellectual disabilities, progressive skeletal sclerosis, cutis laxa, dental enamel dysplasia, and abnormal development in skull and fingers1. Additional cases have been subsequently reported with similar clinical presentations, but some of the symptoms are not equally present in all patients2-6.
What could be the molecular mechanisms underlying the pleiotropic defects seen in LMS patients? It took a long time to begin to answer this question and only with the arrival of the genomic era have the tools become available to search for possible underlying genetic alterations. Sousa and colleagues have conducted genomic analysis with DNA obtained from five LMS-affected individuals7. Remarkably, whole-exome sequencing data have identified mutations in one allele of the PTDSS1 gene in all affected individuals. Three cases showed heterozygous mutations yielding a 1058A>G substitution, generating an amino acid change of Q353R. One individual was heterozygous for an 805C>T substitution causing a P269S change, while another case showed heterozygosity for a 794T>C mutation that lead to a L265P mutation. The PTDSS1 gene encodes for PSS1, one of two phosphatidylserine (PS) synthesizing enzymes that is critical for PS biosynthesis in mammalian cells. PSS1 catalyzes the head-group exchange using phosphatidylcholine (PC) as substrate, while PSS2 uses phosphatidylethanolamine (PE)8,9 (Fig. 1A). Both PSS1 and PSS2 are multi-membrane- spanning proteins located in the ER and structure predictions suggest that they possess nine transmembrane domains with the N-terminus facing the cytoplasm (Fig. 1B). This putative topology places the LMS mutations identified by Sousa and colleagues to the ER-luminal side.
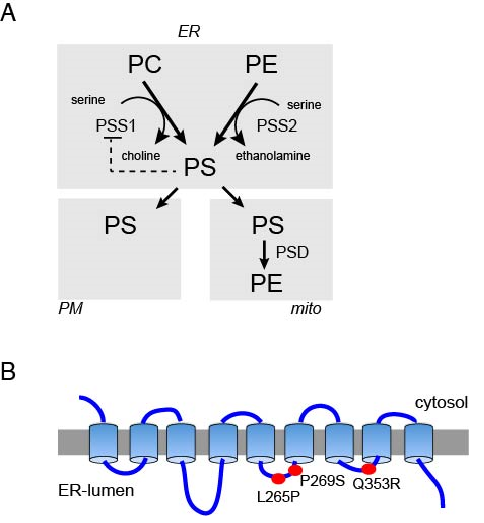
Figure 1: (A) The metabolic routes of phosphatidylserine (PS) synthesis and metabolism. PS is synthesized from PC and PE by headgroup exchange reactions catalyzed by PSS1 and PSS2 enzymes, respectively. PSS1 shows strong product feed-back inhibition. It is less clear if PSS2 has a similar regulation. PS is transported out of the ER to the PM and mitochondria and possibly other membranes by non-vesicular lipid transport. PS may also leave the ER by vesicular transport reaching other membrane compartments (not shown). (B) The putative topology of the PSS1 enzyme showing that the indicated disease-causing mutations are located in the ER luminal side of the protein.
PS makes up about 10-20% of total cellular phospholipids, the majority located in the inner leaflet of PM and the luminal side of the ER but in mammalian cells some PS can also be found in endosomes9-12. PS is responsible for the majority of the acidic character of the inner leaflet of PM11 and it was shown to be important for membrane recruitment and regulation of several signaling proteins including PKC13,14, PLC15 and Ras/Raf16,17 just to name a few. Externalization of PS from inner to outer leaflet of the PM is an early sign of apoptosis and is also critical for platelet aggregation and blot clotting18. How PS gets from its site of synthesis in the ER to the PM has been a question not fully understood. It has been assumed that the majority of PS gets to the PM via vesicular membrane trafficking18-20. Only recently has it become apparent that non-vesicular transfer between the ER and the PM represents a mechanism that can also be important for PS delivery to the PM21-23. It is of great importance that PS synthesis is under a strong product inhibition24. Therefore, the discovery that PSS1 mutants identified in LMS by Sousa and colleagues7 are insensitive to PS-mediated inhibition has explained the dominant inheritance of the disease. LMS mutations reproducibly mimicked the craniofacial defects observed in LMS patients in a zebrafish model, showing that this mutation is sufficient to explain the developmental defects observed in this disease7.
However, now the question arose how PS overproduction leads to the problems seen in LMS? While answers to this question will not be found for a while, our recent findings have shed some light on the complexity of lipid changes caused by the PSS1 mutations. In search of a function of PI4KA, one of PI 4-kinase enzymes that generate PI4P in the PM, we found that PS levels were reduced by 50% when cells were incubated in the presence of an inhibitor of the PI4KA enzyme for 24-36 h. Notably, a similar reduction in total PS amount was observed in cells in which PI4KA was genetically inactivated23. We also observed that this reduction was due to rapid inhibition of PS synthesis by the PI4KA inhibitor. Since the activity of the PSS1 enzyme assayed in isolated membranes was insensitive to the inhibitor, we reasoned that inhibition of PS synthesis was indirect and perhaps due to a defect in the transport of PS out of the ER. Together with the strong PS-mediated feed-back on PS synthesis, a PS transport defect could explain the inhibition of PS synthesis. Indeed, cells transfected with LMS mutant PSS1 enzymes showed massive PS synthesis that was no more sensitive to inhibition of PI4KA. Importantly, in these cells the PS reporter based on the Lactadherin C2 domain (Lact-C2)11decorated the ER, suggesting accumulation of PS at least partly on the cytoplasmic leaflet of the ER25.
These observations were consistent with recent data showing that PI4P gradient between the PM and the ER drives the non-vesicular transport of PS from the ER to the PM21-23. This transport is carried out by ORP5/8 proteins in mammalian cells and by Osh6p in yeast. Such PI4P gradient-driven transport of cholesterol from the ER to the Golgi has already been established26,27. The PI4P gradient is generated by PI4KA at the PM or PI4KB in the Golgi and the Sac1 phosphatase in the ER opposing the PM and the Golgi at membrane contact sites28. The tight connection between PI4P generation at the PM and PS transport raised the question whether uncontrolled production of PS would affect PI4P metabolism. This question was especially relevant in light of the findings that Sac1 phosphatase activity is strongly stimulated by PS29. Indeed, we found that LMS mutant PSS1 overexpression increased Sac1 activity and not only decreased PI4P levels in the plasma membrane (PM), but it also decreased PI4P in the Golgi. These PI4P changes could affect non-vesicular transport of any lipid that relies upon PI4P gradients (Fig. 2).
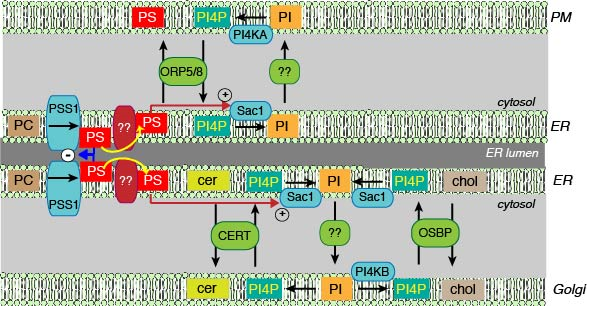
Figure 2: Schematic cartoon showing the relationship between PI4P gradients and non-vesicular lipid transport and the central role of PS in controlling these processes. PI is synthesized in the ER and is phosphorylated to PI4P by one of four distinct PI4K enzymes in the PM, Golgi and endosomes (the last one is not shown for simplicity). How PI gets from the ER to these respective compartments and presented to the PI4Ks is not fully understood. Conversely, the Sac1 phosphatase can dephosphorylate PI4P to PI in the ER. It is a matter of debate whether Sac1 can access PI4P in “trans” in contact sites where the ER and the respective membranes are kept in close proximity or it only dephosphorylates PI4P delivered to the ER by lipid transfer proteins, such as OSBP or ORPs from the adjacent membranes. OSBP and some ORPs can bind and transfer lipids, such as cholesterol (for OSBP) or PS (ORP5/8) as well as PI4P using the same lipid binding cavity.Therefore, the PI4P gradient can be used to drive the directional transport of cholesterol from the ER to the Golgi or PS from the ER to the PM. PI4P in the Golgi is also essential for the transport of ceramide from the ER to the Golgi by CERT, although the ability of the lipid binding domain of CERT to bind PI4P has not been documented yet. The unique position of PS to regulate the PI4P gradients is due to its ability to activate the Sac1 phosphatase, thereby affecting the PI4P gradient regardless of the compartment (PM, Golgi and probably endosomes as well). The other important point of regulation is the negative feed-back of PS levels in the ER on the PSS1 enzyme. Once this feed-back regulation is lost, as in LMS, the excess PS unleashes an avalanche of event by activating Sac1.
These data showing a strong reciprocal relationship between PI4P gradients between the PM and the ER and PS metabolism could explain major problems in lipid metabolism and PI lipid signaling during development. Yet, it is striking that hyperostosis is the leading (although not exclusive) or most noticeable feature of this disease. What does PS have to do with bone metabolism? This question has also been investigated and studies have reported that membrane encapsulated vesicles of 50-200 nm called “matrix vesicles” are critical for mineral deposition in newly forming bones30. These vesicles are enriched in PS, a lipid with strong Ca2+binding property, which plays a role in early stages of mineralization31. Osteoclast differentiation and function have also been found depressed by injected PS liposomes32. It is most likely that excess PS produced by cells at sites of bone formation and maintenance is secreted into the matrix profoundly changing the balance between bone deposition and resorption. How PS gets out of these cells and how other organs such as the brain are affected are important questions and subjects of future investigations.
Concluding Remarks
There are a number of questions still to be understood about the way cells handle PS at normal or extreme PS producing conditions. Importantly, our studies showed a decreased rather than the expected increase in PM PS levels in cells expressing the PSS1 mutant enzymes25. This may be due to an enhanced flipping of PS from the inner to outer leaflet by scramblases as suggested by the large increase in Annexin V positivity of cells obtained from LMS patients7. Also, we do not have a good understanding of the distribution of PS between the two leaflets of the ER. We found accumulation of PS on the cytosolic leaflet of the ER in PSS1 mutant expressing cells, and clearly, PS can only affect Sac1 function from the cytoplasmic side. Yet, if the putative topology of the PSS1 enzyme is correct, the LMS mutations that abolish PS sensing are in the luminal side (Fig. 1B). We can assume that PS is also increased in the luminal side of the ER membrane. How PS moves between the two leaflets in the ER (or Golgi) membrane is still a matter of investigations18. Another important question to be studied is the relative contribution of vesicular and non-vesicular transport of PS from the ER to the PM. How does the extra PS in the ER in LMS patients affect mitochondrial function? We found that PSS1 mutant overexpression also increased the amount of PE presumably formed from PS in the mitochondria through decarboxylation. Does the extra PE in the mitochondria also affect mitochondrial functions?
So clearly we have more questions than answers. Identification of the mutations that cause LMS was a major development. It synergized with other discoveries related to PS metabolism20 and tool development, such as the introduction of Lact-C2 to follow PS distribution in living cells11. The importance of PS in the inner leaflet of PM in the recruitment and regulation of many key signaling nodes (PLC, PKC, Ras/Raf) suggests that disturbances in PS trafficking can have far reaching consequences via affecting early signaling events. Compounding on these defects are the cholesterol and sphingolipid changes due to the effects of PS overproduction on PI4P gradients between the Golgi and ER. It will take some time to sort out to what extents these changes contribute to the developmental defects and what genetic modifiers affect the severity of the various organ dysfunctions. Although LMS is a rare disease better understanding the underlying biochemical changes will greatly advance our knowledge on several aspects of cellular signaling and lipid metabolism. This knowledge would then deepen or insights into many other pathological conditions beyond LMS and identify potential new targets for therapeutic interventions.
Acknowledgement
The research of TB and MS is supported by the intramural research program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health.
References
- Lenz WD, Majewski F. A generalized disorders of the connective tissues with progeria, choanal atresia symphalangism hypoplasia of dentine and craniodiaphyseal hypostosis. Birth Defects Orig Artic Ser. 1974; 10(12): 133-6.
- Robinow M, AJ Johanson, TH Smith. The Lenz Majewski hyperostotic dwarfism A syndrome of multiple congenital anomalies mental retardation and progressive skeletal sclerosis. J Pediatr. 1977; 91(3): 417-21.
- Dateki S, Kondoh T, Nishimura G. A Japanese patient with a mild Lenz Majewski syndrome. J Hum Genet. 2007; 52(8): 686-9.
- Chrzanowsk KH, Fryns JP, Krajewska Walasek M. Skeletal dysplasia syndrome with progeroid appearance characteristic facial and limb anomalies multiple synostoses and distinct skeletal changes a variant example of the Lenz Majewski syndrome. Am J Med Genet. 1989; 32(4): 470-4.
- Wattanasirichaigoon D, Visudtibhan A, Jaovisidha S. Expanding the phenotypic spectrum of Lenz Majewski syndrome facial palsy cleft palate and hydrocephalus. Clin Dysmorphol. 2004; 13(3): 137-42.
- Tamhankar PM, Vasudevan L, Bansal V. Lenz Majewski syndrome Report of a case with novel mutation in PTDSS1 gene. Eur J Med Genet. 2015; 58(8): 392-9.
- Sousa SB, Jenkins D, Chanudet E. Gain of function mutations in the phosphatidylserine synthase 1 (PTDSS1) gene cause Lenz Majewski syndrome. Nat Genet. 2014; 46(1): 70-6.
- Tomohiro S, Kawaguti A, Kawabe Y. Purification and characterization of human phosphatidylserine synthases 1 and 2. Biochem J. 2009; 418(2): 421-9.
- Vance JE, Tasseva G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta. 2013; 1831(3): 543-54.
- Higgins JA, Dawson RM. Asymmetry of the phospholipid bilayer of rat liver endoplasmic reticulum. Biochim Biophys Acta. 1977; 470(3): 342-56.
- Yeung T, Gilbert GE, Shi J. Membrane phosphatidylserine regulates surface charge and protein localization. Science. 2008; 319(5860): 210-3.
- Fairn GD, Schieber NL, Ariotti N. High resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J Cell Biol. 2011; 194(2): 257-75.
- Stahelin RV, Rafter JD, Das S.The molecular basis of differential subcellular localization of C2 domains of protein kinase C alpha and group IVa cytosolic phospholipase A2. J Biol Chem. 2003; 278(14): 12452-60.
- Conesa Zamora P, Lopez Andreo MJ, Gómez Fernández JC. Identification of the phosphatidylserine binding site in the C2 domain that is important for PKC alpha activation and in vivo cell localization. Biochemistry. 2001; 40(46): 13898-905.
- Lomasney JW, Cheng HF, Roffler SR. Activation of phospholipase C delta1 through C2 domain by a Ca(2+) enzyme phosphatidylserine ternary complex. J Biol Chem. 1999; 274(31): 21995-2001.
- McPherson RA, Harding A, Roy S. Interaction of c Raf 1 with phosphatidylserine and 14- 3-3. Oncogene. 1999; 18: 3862-3869.
- Cho KJ, Park JH, Piggott AM. Staurosporines disrupt phosphatidylserine trafficking and mislocalize Ras proteins. J Biol Chem. 2012; 287(52): 43573-84.
- Hankins HM, Baldridge RD, Xu P. Role of flippases scramblases and transfer proteins in phosphatidylserine subcellular distribution. Traffic. 2015; 16(1): 35-47.
- Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010; 39: 407-27.
- Kay JG, Koivusalo M, Ma X. Phosphatidylserine dynamics in cellular membranes. Mol Biol Cell. 2012; 23(11): 2198-212.
- Maeda K, Anand K, Chiapparino A. Interactome map uncovers phosphatidylserine transport by oxysterol binding proteins. Nature. 2013; 501(7466): 257-61.
- Moser von Filseck J, ?opi? A, Delfosse V. INTRACELLULAR TRANSPORT. Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4 phosphate. Science. 2015; 349(6246): 432-6.
- Chung J, Torta F, Masai K. Intracellular transport. PI4P/phosphatidylserine countertransport at ORP5 and ORP8 mediated ER plasma membrane contacts. Science. 2015; 349(6246): 428-32.
- Kuge O, Hasegawa K, Saito K. Control of phosphatidylserine biosynthesis through phosphatidylserine mediated inhibition of phosphatidylserine synthase I in Chinese hamster ovary cells. Proc Natl Acad Sci U S A. 1998; 95(8): 4199- 203.
- Sohn M, Ivanova P, Brown HA. Lenz Majewski mutations in PTDSS1 affect phosphatidylinositol 4 phosphate metabolism at ER PM and ER Golgi junctions. Proc Natl Acad Sci U S A. 2016; 113(16): 4314-9.
- de Saint Jean M, Delfosse V, Douguet D. Osh4p exchanges sterols for phosphatidylinositol 4 phosphate between lipid bilayers. J Cell Biol. 2011; 195(6): 965-78.
- Mesmin B, Bigay J, Moser von Filseck J. A four step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER Golgi tether OSBP. Cell. 2013; 155(4): 830-43.
- Moser von Filseck J, Drin G. Running up that hill: How to create cellular lipid gradients by lipid counter flows. Biochimie. 2016; 130: 115-121.
- Zhong S, Hsu F, Stefan CJ. Allosteric activation of the phosphoinositide phosphatase Sac1 by anionic phospholipids. Biochemistry. 2012; 51(15): 3170-7.
- Merolli A, Santin M. Role of phosphatidyl serine in bone repair and its technological exploitation. Molecules. 2009; 14(12): 5367-81.
- Wu LN, Genge BR, Wuthier RE. Analysis and molecular modeling of the formation structure and activity of the phosphatidylserine calcium phosphate complex associated with biomineralization. J Biol Chem. 2008; 283(7): 3827-38.
- Wu Z, Ma HM, Kukita T. Phosphatidylserine containing liposomes inhibit the differentiation of osteoclasts and trabecular bone loss. J Immunol. 2010; 184(6): 3191-201.