
Mini-review on current strategies to knockdown long non-coding RNAs
Kim A Lennox and Mark A Behlke*
Integrated DNA Technologies, Inc., Coralville, IA, 52241, USA
Text
The discovery of functional long non-coding RNAs (lncRNAs) has challenged the paradigm that most RNAs encode proteins. LncRNAs are an abundant class of RNAs >200 nt in length that were previously assumed to be transcriptional noise. It is now widely accepted that some of these lncRNAs have diverse regulatory roles in gene expression, can serve as structural components for shaping nuclear organization and can even act as decoys for other RNAs or proteins1-3. While tens of thousands of lncRNA transcripts have been cataloged, the biological function of the majority of lncRNAs still remains a mystery. Determining the individual functionality of members of this recently discovered class of RNAs is important for better understanding of developmental biology, evolution and genetic disease.
Similar to proteins, individual lncRNAs have specific subcellular distributions that are critical for their function4. Some lncRNAs are enriched in the nucleus and are involved in regulating nuclear processes such as transcription and RNA processing. Other lncRNAs are enriched in the cytoplasm where they can impact protein localization or modulate mRNA stability and translation. Some lncRNAs are more equally distributed between the nucleus and the cytoplasm. The location of the primary reservoir of a lncRNA can impact the ability to knock down expression levels of that species.
Three strategies are commonly used to knockdown or knockout lncRNAs, including: degradation of the RNA by RNA interference (RNAi), degradation of the RNA by RNase H activate antisense oligonucleotides (ASOs), or gross deletion/alteration at the DNA level using CRISPR/Cas9 genome editing methods (Figure 1). Each of these tools have their own advantages and disadvantages, and success can be influenced by the subcellular localization of the lncRNA and the transcriptional landscape within which it resides.
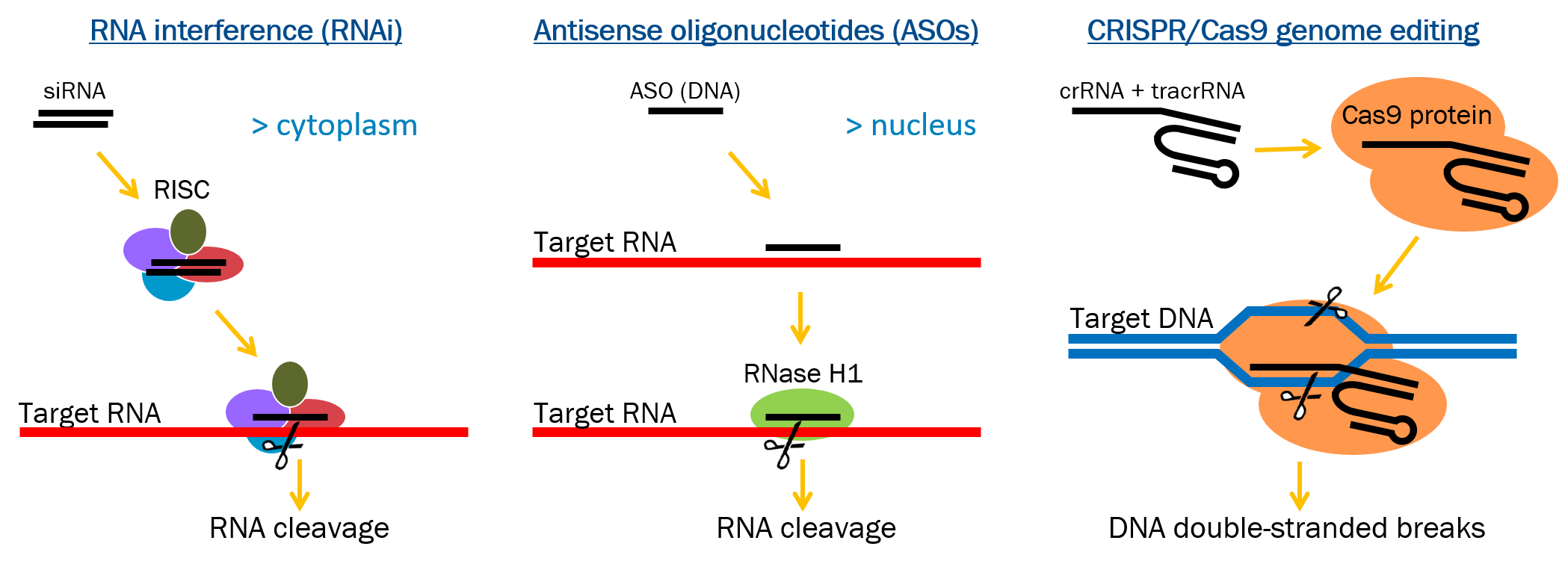
Figure 1: Schematic of the three common methods used to knockdown or knockout lncRNAs. RNA interference (RNAi) utilizes the multi-protein RNAi-induced silencing complex (RISC) containing a siRNA to specifically degrade the targeted RNA. Antisense oligonucleotides (ASOs) bind to their targeted RNA and use endogenous RNase H1, an enzyme that cleaves the RNA in an RNA/DNA heteroduplex. CRISPR-Cas9 genome editing makes alterations at the genomic level by using a target specific crRNA hybridized to the tracrRNA, which is complexed to the Cas9 protein.
RNAi is a commonly employed knockdown technique that utilizes the multi-protein RNAi-induced silencing complex (RISC) to suppress mRNAs5. The human RISC loading complex (RLC), is comprised of three proteins (Dicer, TRBP and Ago2) responsible for processing longer dsRNAs into the mature siRNAs and loading these siRNAs into Ago2. It has previously been demonstrated that RNAi-mediated mRNA degradation occurs in the cytoplasm6. In a series of fractionation and co-immunoprecipitation experiments, Stalder et al. more specifically demonstrate that canonical RISC loading, subsequent binding with its targeted mRNA and Ago2-mediated cleavage of the mRNA occurs primarily at the rough endoplasmic reticulum, where mRNAs are translated into proteins7. RNase H-mediated antisense RNA knockdown capitalizes on the endogenous RNase H1 enzyme, which is most abundant in the nucleus where it is thought to function in DNA replication and repair8-11. Alternatively, steric blocking ASOs can be used to block splice junctions to reduce accumulation of mature lncRNA transcripts or block access to key functional domains without triggering degradation of the target RNA12,13. Steric blocking ASOs are made of chemically modified residues that do not support RNase H1 cleavage, such as 2’-modified ribose or morpholino backbones14. Both RNAi and RNase H-active ASOs rely upon naturally present effector molecules to degrade the lncRNA. In contrast, CRISPR/Cas9 genome editing methods rely upon a bacterial endonuclease enzyme that can be targeted to desired sites in the genome by a site-specific guide RNA where it generates double-stranded DNA breaks at or around the target site15,16. The cellular repair machinery heals the double-stranded breaks, leaving small “scars” in the genome, or can even be used to delete large blocks of DNA and thereby eliminate the lncRNA at the genomic level. CRISPR/Cas9 methods can also be used to introduce new sequences at the target loci, such as transcriptional terminators that will prevent production of full-length lncRNAs. Understanding the nature of the lncRNA of interest can better assist with selecting the optimal knockdown technique.
With an extensive background of mRNA knockdown experience, we expected that applying the same repertoire of reagents to suppress lncRNAs would be easily accomplished. However, we found that some lncRNAs were difficult to suppress, and eventually noticed what seemed to be a correlation that RNAi-based methods (small interfering RNAs, siRNAs; or Dicer substrate RNAs, DsiRNAs) were less effective against nuclear-localized lncRNA targets. We hypothesized that these nuclear lncRNAs would be more easily suppressed using RNase-H mediated antisense knockdown, since RNase H is predominantly found in the nucleus. Conversely, we thought RNAi, a mostly cytoplasmic process, might be more effective when targeting cytoplasmic populations of lncRNAs. To test this possibility, a systematic study was performed in cell culture comparing the effectiveness of ASOs (20mer DNA-PS, 20mer 2’OMe-PS gapmers or 16mer LNA-PS gapmers (Exiqon), where PS = phosphorothioate linkages, LNA = locked nucleic acid and 2’OMe = 2’-O-Methyl RNA) or RNAi (DsiRNAs, siRNA, or Silencer® Select siRNAs (Thermo Fisher Scientific)) against seven lncRNAs with variable localization patterns17. Nuclear lncRNAs (MALAT1 and NEAT1), cytoplasmic lncRNAs (DANCR and OIP5-AS1) or dual-localized lncRNAs (TUG1, CasC7 and HOTAIR) were targeted by either ASOs (18 sites/target) or RNAi (28 sites/target). As there is a wide range of efficacy between sites depending on parameters such as GC content and target accessibility, sites were selected by using optimized design algorithms (RNAi) or using well-defined stringent in-house design criteria (ASOs) to maximize the probability of comparing highly active sites between to the two knockdown strategies. The results were consistent with our hypothesis: nuclear lncRNAs were more easily knocked down when using ASOs, while cytoplasmic targets were more easily knocked down when using RNAi (Figure 2). When comparing the potency of the various ASO chemical modification strategies, it was found that while the DNA-PS and 2’OMe-PS ASOs had very similar knockdown patterns between the different sites, the 2’OMe-PS gapmers were consistently more potent. This was expected, as the 2’OMe bases increase the binding affinity of the ASO to the target. The 2’OMe-PS gapmer and LNA-PS gapmer ASOs typically showed similar potency at the 10 nM dose; however, the LNA-PS gapmers were often more potent at lower doses. There was no overall difference in activity between any of the various RNAi reagents tested. Duration of knockdown was not addressed in this study; suppression lasting 3-7 days is typically seen in cell culture (where dilution from cell division can be limiting by reducing concentration of the ASOs or siRNAs), whereas knockdown for several weeks or even a month can be seen in non-dividing cells in vivo18,19. While this study only examined lncRNA knockdown in cell culture, similar relative activities between the different reagent classes would be expected in vivo if suitable delivery tools were employed. For in vivo use, a clear performance advantage would be predicted for the short LNA-PS gapmers if administered by naked IV injection (without any delivery aid)20. Use of a delivery tool, such as a lipid nanoparticle (LNP) or conjugated ligand (such as cholesterol or an antibody) can improve delivery of ASOs and would be required for use of any RNAi reagents in vivo18,19,21,22.
So far, we have commented on the “relative ease” of either knockdown technique to suppress their targets based on whether the lncRNA was predominantly nuclear or cytoplasmic. A closer look at the data shows that ASOs are also effective at degrading cytoplasmic RNAs (Figure 2). ASOs not only have the innate ability to target nuclear-retained RNAs, but they can also degrade nascent RNAs prior to cytoplasmic exportation. Also, a recent study examining the rate-limiting step of mRNA degradation by ASOs found that RNase H1 was also present in low levels in the cytoplasm23. While ASO-mediated mRNA degradation occurred more rapidly in the nucleus, it also occurred at a slower rate in the cytoplasm. This suggests that if the localization of the lncRNA is unknown, selecting ASO knockdown (as opposed to RNAi) would have the best chance of success. Interestingly, our study showed that when targeting dual-localized lncRNAs, improved knockdown could be achieved by combining ASOs and siRNAs together. This was predictable, considering that the ASO could most rapidly degrade the nuclear lncRNA population and the siRNA could most rapidly degrade the cytoplasmic lncRNA population.
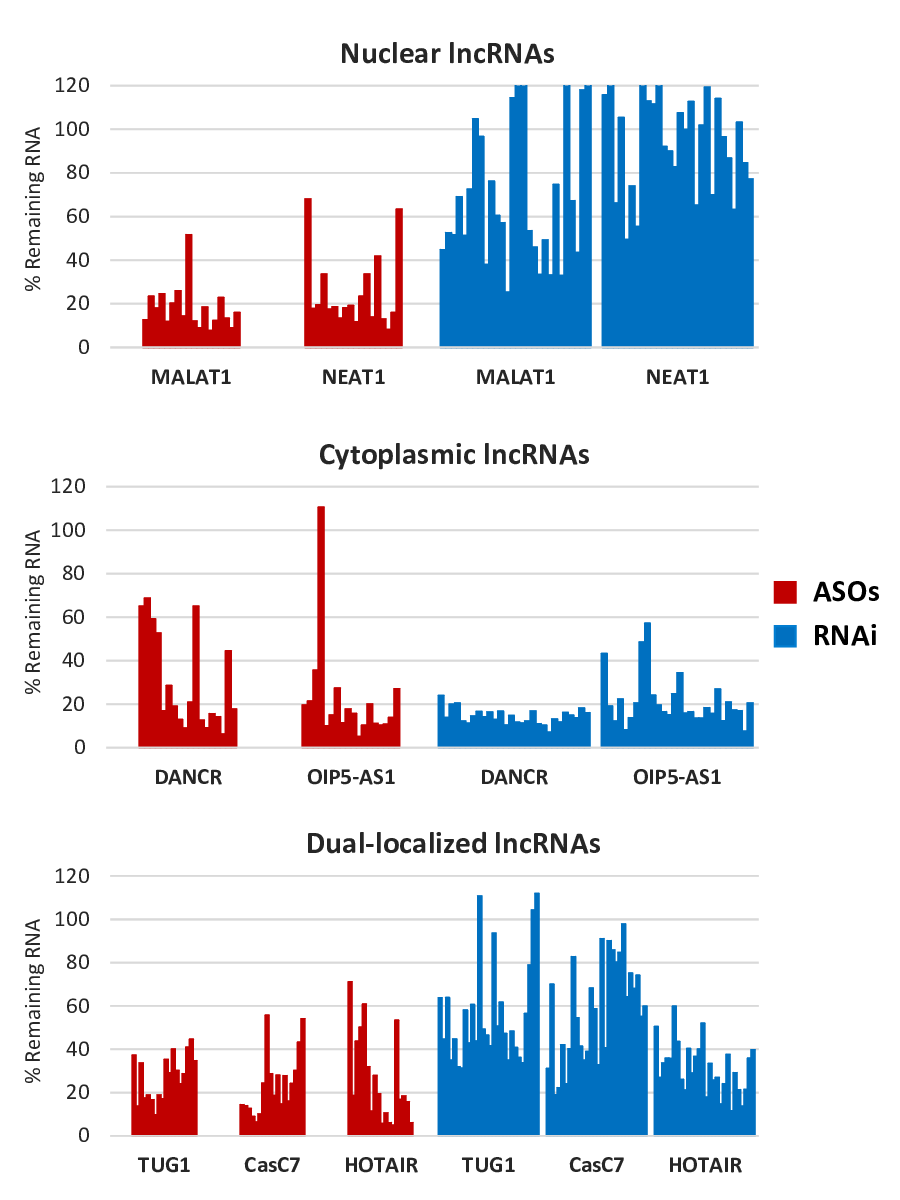
Figure 2: Overview of lncRNA knockdown results comparing method efficacy with lncRNA subcellular localization (17). Antisense oligonucleotides (LNA-PS gapmers, 2’OMe-PS gapmers or DNA-PS) or RNAi reagents (siRNAs, DsiRNAs or Silencer® Select siRNAs) were transfected into HeLa cells with Lipofectamine® 2000 for 24 h at 10 nM dose. ASOs = antisense oligonucleotides; PS = phosphorothioate linkages.
Some lncRNAs will be refractory to knockdown by either ASOs or RNAi. This could occur if the subcellular localization of the lncRNA is not accessible to either RNase H or the RNAi machinery. It could also occur if the lncRNA is highly structured or blocked due to excessive protein binding or hybridizing to other cellular nucleic acids. We tested a wide variety of ASOs and RNAi reagents against NRON, a lncRNA that functions as a protein sponge, and were unable to achieve high levels of knockdown (unpublished results). In these circumstances, using a technique such as CRISPR/Cas9 genome editing might be necessary. Using a genome editing tool to knockout a lncRNA is not trivial, as making a small indel in the targeted lncRNA most likely will not disrupt transcription; no ‘open reading frame’ exists that is easily disrupted by out-of-frame indel events. Transcribed lncRNAs containing newly introduced indels may still retain functional domains and/or binding sites that would obscure phenotypic analysis. A couple of strategies have successfully employed CRISPR/Cas9 genome editing to knockout lncRNAs. One strategy uses dual guide RNAs to simultaneously break the DNA at two specific locations, excising out a large fragment of the genomic loci that encodes the lncRNA. Proof-of-concept for this technique successfully knocked out lncRNA-21A, UCA1 and AK023948 by removing large fragments ranging from 475 bp (lnRNA-21A) to 5.6 kb (UCA1)24. A second strategy uses CRISPR/Cas9 to engineer an RNA destabilizing element (such as a poly(A) signal, a miRNA binding site, a self-cleaving ribozyme, etc.) into the genomic sequence, causing increased lncRNA transcript turnover/degradation. Using this strategy initially with zinc fingers nucleases, Gutschner et al developed a MALAT1 knockout model by integrating a poly(A) signal upstream of the transcriptional start site, yielding a >1000 fold reduction of MALAT1 levels25,26. The advent of the CRISPR/Cas9 technology greatly simplified this approach. CRISPR-inhibition (CRISPRi) is a technique that either uses a catalytically inactive (dead) Cas9 (dCas9) protein to block RNA polymerase function, or, if higher transcriptional repression is desired, couples the dCas9 with a transcriptional repressor (such as KRAB). Gilbert et al targeted six different lncRNAs using dCas9-KRAB, achieving >80% knockdown for 5/6 lncRNAs27. In particular, effective use of CRISPRi methods requires that the location of enhancer/promotor elements are known and also if these regulatory elements solely control expression of the lncRNA or also contribute to expression of other (coding) transcripts. Using any CRISPR method, transcripts that overlap, are transcribed from the opposite strand, or are further processed into different isoforms could also inadvertently be knocked out, confounding data interpretation. In fact, Goyal et al recently demonstrated that 62% of the 15,929 lncRNAs analyzed in their study were poor targets for CRISPR/Cas9 genome editing due to containing bidirectional or internal promoters; thus, their knockout could potentially affect neighboring genes. The authors selected a few examples from this analysis, and demonstrated that using CRISPRi to knockout lncRNAs with loci that overlap other genes did indeed affect their neighboring genes. In contrast, RNAi or ASO knockdown of these same lncRNA targets was specific to the targeted transcript28.
All techniques that suppress gene expression have a risk of off-target effects (OTEs), where related genes that have sequence homology to the intended target are suppressed by cross-hybridization with the targeting nucleic acids. The specificity of ASOs relies solely upon Watson-Crick nucleic acid hybridization and OTEs are easier to predict by bioinformatic analysis than other methods; these reagents can show single-base specificity, especially when using high affinity short ASOs29. While RNAi methods can show single-base specificity, these methods have a higher risk of unsuspected OTEs due to crosstalk with miRNA pathways, where a short 6-7 base “seed region” defines specificity of suppression, giving rise to a risk of affecting many unrelated genes30. However, a large element of this OTE pathway derives from translational suppression, which, by definition, cannot directly affect lncRNA function. CRISPR methods likewise have a risk of OTEs. Use of direct ribonucleoprotein (RNP) gene editing methods (where Cas9 protein and guide RNAs alone are employed in a DNA-free format) show far lower OTE profiles than methods that employ plasmid-based overexpression of the Cas9 and guide RNAs, somewhat mitigating the OTEs risk31. Further, Cas9 mutants have been developed that have a significantly reduced risk of acting at off-target sites32. Hence, use of improved ‘newer generation CRISPR methods’ can lower the risk of an OTE affecting results. As an additional concern, CRISPR methods that permanently alter the DNA of a cell often result in the creation of different mutations on each chromosome in a cell or between cells in a population. It may be necessary to isolate clonally pure cell lines for study before definitive conclusions can be made from these kinds of genome editing experiments.
The discovery of thousands of lncRNAs and their ambiguous roles in cellular processes has increased the need to have reliable knockdown techniques to help determine their individual functions. Being able to select the knockdown method most likely to give the best results saves time and money, and further progresses lncRNA research. Here we described a repertoire of techniques that can be used depending on lncRNA localization, lncRNA transcript (in)accessibility to enzymes, and the transcriptional landscape within which they reside.
References
- Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016; 17: 47-62.
- Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013; 154: 26-46.
- Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013; 14: 699-712.
- Chen LL. Linking Long Noncoding RNA Localization and Function. Trends Biochem Sci. 2016; 41: 761-772.
- Dorsett Y, Tuschl T. siRNAs: applications in functional genomics and potential as therapeutics. Nat Rev Drug Discov. 2004; 3: 318-329.
- Zeng Y, Cullen BR. RNA interference in human cells is restricted to the cytoplasm. RNA. 2002; 8: 855-860.
- Stalder L, Heusermann W, Sokol L, et al. The rough endoplasmatic reticulum is a central nucleation site of siRNA-mediated RNA silencing. EMBO J. 2013; 32: 1115-1127.
- Wu H, Lima WF, Zhang H, et al. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J Biol Chem. 2004; 279: 17181-17189.
- Vickers TA, Koo S, Bennett CF, et al. Efficient reduction of target RNAs by small interfering RNA and RNase H-dependent antisense agents. A comparative analysis. J Biol Chem. 2003; 278: 7108-7118.
- Suzuki Y, Holmes JB, Cerritelli SM, et al. An upstream open reading frame and the context of the two AUG codons affect the abundance of mitochondrial and nuclear RNase H1. Mol Cell Biol. 2010; 30: 5123-5134.
- Lima WF, Wu H, Crooke ST. CRC Press, Boca Raton. 2008; 2 ed: 47-74.
- Kurian L, Aguirre A, Sancho-Martinez I, et al. Identification of novel long noncoding RNAs underlying vertebrate cardiovascular development. Circulation. 2015; 131: 1278-1290.
- Ulitsky I, Shkumatava A, Jan CH, et al. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011; 147: 1537-1550.
- Veltrop M, Aartsma-Rus A. Antisense-mediated exon skipping: taking advantage of a trick from Mother Nature to treat rare genetic diseases. Exp Cell Res. 2014; 325: 50-55.
- Wright AV, Nunez JK, Doudna JA. Biology and Applications of CRISPR Systems: Harnessing Nature's Toolbox for Genome Engineering. Cell. 2016; 164: 29-44.
- Barrangou R, Doudna JA. Applications of CRISPR technologies in research and beyond. Nat Biotechnol. 2016; 933-941.
- Lennox KA, Behlke MA. Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res. 2016; 44: 863-877.
- Behlke MA. Progress towards in vivo use of siRNAs. Mol Ther. 2006; 13: 644-670.
- Rettig GR, Behlke MA. Progress toward in vivo use of siRNAs-II. Mol Ther. 2012; 20: 483-512.
- Straarup EM, Fisker N, Hedtjarn M, et al. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res. 2010; 38: 7100-7111.
- Prakash TP, Lima WF, Murray HM, et al. Lipid nanoparticles improve activity of single-stranded siRNA and gapmer antisense oligonucleotides in animals. ACS Chem Biol. 2013; 8: 1402-1406.
- Prakash TP, Graham MJ, Yu J, et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014; 42: 8796-8807.
- Vickers TA, Crooke ST. The rates of the major steps in the molecular mechanism of RNase H1-dependent antisense oligonucleotide induced degradation of RNA. Nucleic Acids Res. 2015.
- Ho TT, Zhou N, Huang J, et al. Targeting non-coding RNAs with the CRISPR/Cas9 system in human cell lines. Nucleic Acids Res. 2015; 43: e17.
- Gutschner T, Baas M, Diederichs S. Noncoding RNA gene silencing through genomic integration of RNA destabilizing elements using zinc finger nucleases. Genome Res. 2011; 21: 1944-1954.
- Gutschner T, Hammerle M, Eissmann M, et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013; 73: 1180-1189.
- Gilbert LA, Horlbeck MA, Adamson B, et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014; 159, 647-661.
- Goyal A, Myacheva K, Gross M, et al. Challenges of CRISPR/Cas9 applications for long non-coding RNA genes. Nucleic Acids Res. 2016.
- Carroll JB, Warby SC, Southwell AL, et al. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene / allele-specific silencing of mutant huntingtin. Mol Ther. 2011; 19: 2178-2185.
- Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010; 9: 57-67.
- Kim S, Kim D, Cho SW, et al. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014; 24: 1012-1019.
- Kleinstiver BP, Pattanayak V, Prew MS, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016; 529: 490-495.