
Post ER Quality Control: A Role for UDP-Glucose:Glycoprotein Glucosyl Transferase and p97
Ilaria Fregno1,2,3*, Maurizio Molinari1,2,4*
1Università della Svizzera italiana, CH-6900 Lugano, Switzerland
2Institute for Research in Biomedicine, CH-6500 Bellinzona, Switzerland
3Department of Biology, Swiss Federal Institute of Technology, CH-8093 Zurich, Switzerland
4Ecole Polytechnique Fédérale de Lausanne, School of Life Sciences, CH-1015 Lausanne, Switzerland
Human proteinopathies are diseases caused by the expression of defective gene products. In some cases, these diseases involve the degradation of mutant but otherwise functional proteins by the quality control system of the secretory pathway. Our recent study identified two proteins that play a role in post-endoplasmic reticulum (ER) quality control and are potential targets for therapeutic treatment of several proteinopathies1
Text
Secretory and membrane proteins acquire their native structure in the ER, where molecular chaperones and folding enzymes assist the folding process. Despite the presence of a large number of ER-resident folding factors, the folding process may eventually fail. Non-native features such as exposed hydrophobic patches, unpaired cysteine residues and non-native peptidyl-prolyl bonds may alert the ER quality control machinery to ensure that terminally misfolded polypeptides are not released into the secretory pathway (ER QC, yellow arrow in the schematic). Rather, they are dislocated into the cytosol and ultimately degraded by the ubiquitin-proteasome system in a series of events collectively defined as ER-associated degradation (ERAD, red arrow)2.
Intermediates of folding programs, or individual subunits of oligomeric complexes must also be retained in the ER, but they must be preserved from premature degradation. The presence of ionizable residues in the intramembrane portion is a common feature of many subunits of oligomeric complexes. These residues play an essential role in the biogenesis of complexes such as the T-cell receptor (TCR), the natural killer-cell immunoglobulin-like receptor (KIR) and the NKG2D receptor by promoting retention in the folding compartment until association of the appropriate binding partners3-5. However, mutations introducing ionizable residues in the transmembrane domain of secretory proteins may have deleterious consequences such as the inappropriate selection for degradation of these otherwise functional gene products6,7. For example, Ehlers-Danlos syndrome and idiopathic epilepsy are caused by the inappropriate degradation of the zinc transporter protein ZIP13 and the GABAA receptor, respectively, in which an alanine or glycine to aspartic acid mutation is introduced in the transmembrane portion8-10.
Understanding the molecular details of quality control mechanisms operating in living cells is fundamental for the development of new pharmacological treatments that rescue mutant, but functional polypeptides from degradation and promote their transport to the proper site of activity. This would ameliorate the outcome of several human proteinopathies11,12.
A recent study in our lab led to the identification of a new quality control checkpoint operating in the secretory pathway that is alerted by the presence of ionizable residues in the transmembrane region. The peculiarity of this novel checkpoint is that it intervenes after release of the mutant protein from the ER1.
To monitor the consequences of ionizable transmembrane residues, we constructed four different model polypeptides. The chimeras used in our study were type I membrane proteins where the luminal portion was constituted by a folding defective (Null Hong Kong variant [NHK] of human α1-anti trypsin [α1AT]) or a folding competent (α1AT) ectodomain fused to the C-terminal portion of CD3δ, a subunit of the TCR. The transmembrane region of CD3δ naturally contains an ionizable aspartic acid residue at position 6 (chimeras NHKc and α1ATc), which was mutated into an uncharged alanine residue (chimeras NHKcD6A and α1ATcD6A) (Figure 1).
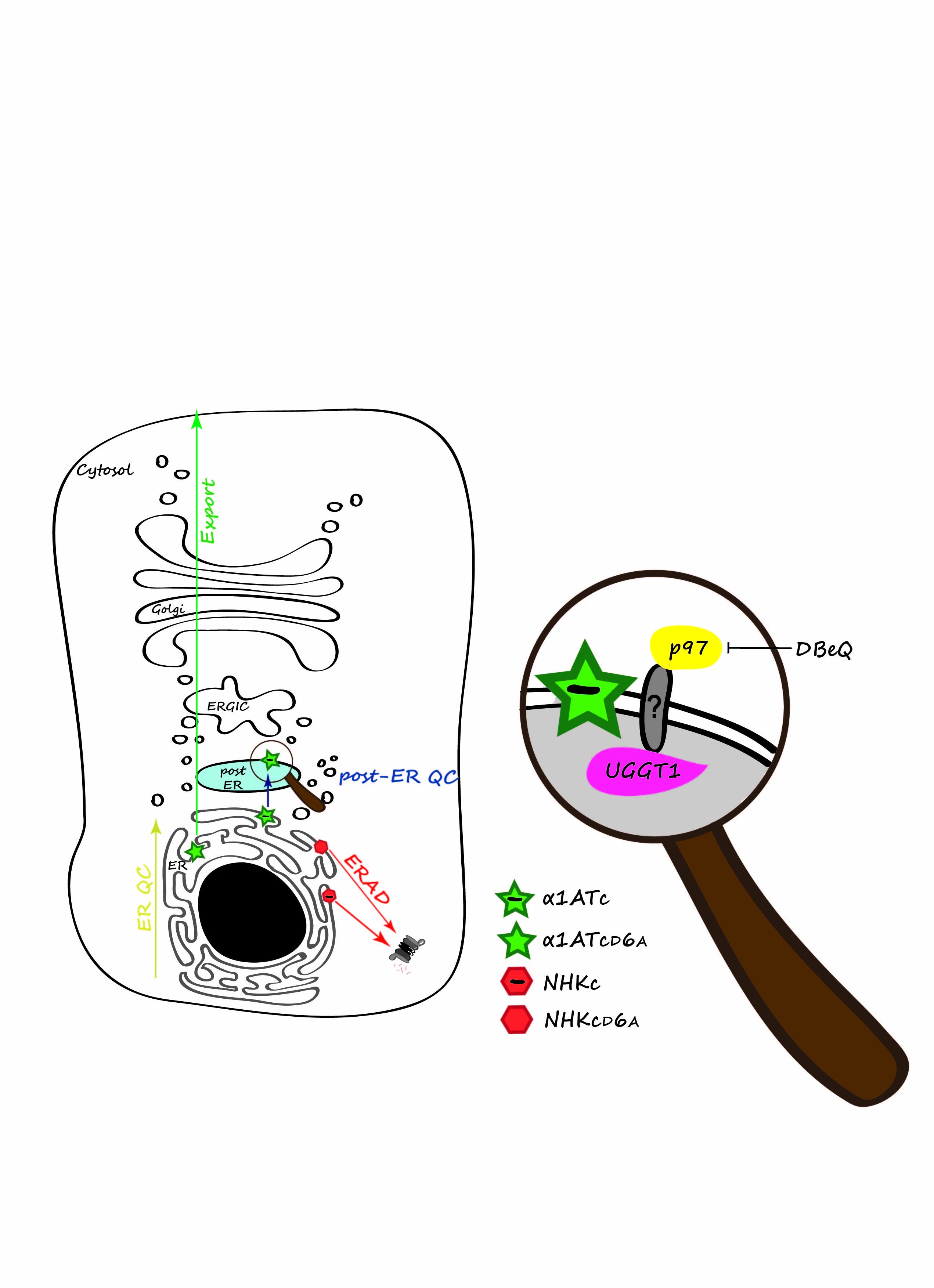
Figure: Protein quality control in mammalian cells. Secretory proteins are folded in the ER. ER QC prevents exit of non-native polypeptides from the ER (yellow arrow). Misfolded proteins are dislocated into the cytosol for proteasomal degradation (red arrow). A second, post-ER QC (blue arrow), prevents secretion of proteins displaying intra-membrane defects. The magnifying glass reveals the p97:UGGT1 post-ER QC complex with the still elusive transmembrane protein linker. Native proteins are exported into the Golgi compartment to their intra- or extra-cellular site of activity (green arrow).
The chimeras displaying the folding-defective ectodomain, NHKc and NHKcD6A, were retained in the ER and were efficiently cleared by the cytosolic proteasome. The presence or absence of the intramembrane ionizable residue made little difference, if at all. This was expected since the ER QC machinery proficiently recognizes misfolded polypeptides and targets them for ERAD. On the other hand, the intramembrane ionizable residue substantially affected the fate of the chimeras displaying a folded ectodomain as the folding-competent chimera lacking the ionizable residue in the intramembrane domain (α1ATcD6A) was efficiently transported to the Golgi, whereas the chimera displaying the intramembrane ionizable residue (α1ATc) was halted in a compartment located between the ER and the Golgi .
The novel, post-ER QC checkpoint relies on the multifunctional, cytosolic AAA ATPase p9713 and UDP-glucose:glycoprotein glucosyl transferase (UGGT1), a luminal ER-resident enzyme responsible for the re-glucosylation of N-linked sugars, a process determining glycopolypeptides retention in the calnexin cycle14. Consistently, pharmacological inhibition of p97 with N2,N4-dibenzylquinazoline-2,4-diamine (DBeQ), a selective ATP-competitive p97 inhibitor15, or individual silencing of p97 and UGGT1 expression restored the Golgi transport of α1ATc. As such, p97 and UGGT1 emerge as novel pharmacologic targets for loss-of-function diseases.
UGGT1 and p97 are soluble proteins of the ER lumen and of the cytosol, respectively. The membrane-bound linker connecting them in a functional post-ER QC complex remains to be identified. The multispanning protein Bap31 is a candidate protein for this function. Bap31 is involved in ERAD and in vesicular transport in the early secretory pathway16,17. Importantly, it has been reported that Bap31 preferentially interacts with charged intramembrane residues18. In our tests, silencing of Bap31 did not rescue the α1ATc trafficking. This could be explained by residual Bap31 that might ensure post-ER retention of α1ATc. Future perspectives include the knockout of Bap31 and other putative components operating in post-ER quality control using a recently developed, powerful gene-editing technology, the CRISPR/Cas9 technology19.
The characterization of mechanisms regulating protein quality control will help in the identification of new targets for pharmacologic treatment of human proteinopathies, i.e., of diseases caused by expression of defective gene products.
Acknowledgement
We thank Giorgia Brambilla Pisoni for helping us in the visual representation of our model. Molinari M. is supported by Signora Alessandra, the Foundation for Research on Neurodegenerative Diseases, the Swiss National Science Foundation (SNF), the Sinergia grant of the SNF and the Comel and Gelu Foundations.
References
- Merulla J, Solda T, and Molinari M. A novel UGGT1 and p97-dependent checkpoint for native ectodomains with ionizable intramembrane residue. Mol Biol Cell, 2015; 26(8): 1532-42.
- Brambilla Pisoni G, Molinari M. Five Questions (with their Answers) on ER-Associated Degradation. Traffic, 2016; 17(4): 341-50.
- Klausner RD, Lippincott-Schwartz J, Bonifacino JS, The T cell antigen receptor: insights into organelle biology. Annu Rev Cell Biol, 1990; 6: 403-31.
- Call ME, Wucherpfennig KW. The T cell receptor: critical role of the membrane environment in receptor assembly and function. Annu Rev Immunol, 2005; 23: 101-25.
- Feng J, Call ME, Wucherpfennig KW. The assembly of diverse immune receptors is focused on a polar membrane-embedded interaction site. PLoS Biol, 2006; 4(5): e142.
- Noack J, Brambilla Pisoni G, Molinari M. Proteostasis: bad news and good news from the endoplasmic reticulum. Swiss Med Wkly, 2014; 144: w14001.
- Hebert DN, Molinari M, In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev, 2007; 87(4): 1377-408.
- Bin BH, Hojyo S, Hosaka T, et al. Molecular pathogenesis of spondylocheirodysplastic Ehlers-Danlos syndrome caused by mutant ZIP13 proteins. EMBO Mol Med, 2014; 6(8): 1028-42.
- Cossette P, Liu L, Brisebois K, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet, 2002; 31(2): 184-9.
- Gallagher MJ, Shen W, Song L, et al. Endoplasmic reticulum retention and associated degradation of a GABAA receptor epilepsy mutation that inserts an aspartate in the M3 transmembrane segment of the alpha1 subunit. J Biol Chem, 2005; 280(45): 37995-8004.
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol, 2003; 4(3): 181-91.
- Romisch K. A cure for traffic jams: small molecule chaperones in the endoplasmic reticulum. Traffic, 2004; 5(11): 815-20.
- Yamanaka K, Sasagawa Y, T. Ogura. Recent advances in p97/VCP/Cdc48 cellular functions. Biochim Biophys Acta, 2012; 1823(1): 130-7.
- Caramelo JJ, Parodi aj. Getting in and out from calnexin/calreticulin cycles. J Biol Chem, 2008; 283(16): p. 10221-5.
- Chou, T.F., et al., Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A, 2011; 108(12): 4834-9.
- Wakana Y, Takai S, Nakajima K, et al. Bap31 is an itinerant protein that moves between the peripheral endoplasmic reticulum (ER) and a juxtanuclear compartment related to ER-associated Degradation. Mol Biol Cell, 2008; 19(5): 1825-36.
- Wang, B, Heath-Engel H, Zhang D, et al. BAP31 interacts with Sec61 translocons and promotes retrotranslocation of CFTRDeltaF508 via the derlin-1 complex. Cell, 2008; 133(6): 1080-92.
- Geiger R, Andritschke D, Friebe S, et al. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat Cell Biol, 2011; 13(11): 1305-14.
- Ledford H. CRISPR: gene editing is just the beginning. Nature, 2016; 531(7593): 156-9.