
Prolactinoma as a Cause of Persistent Hyperprolactinemia in 6-Pyruvoyl-tetrahydropterin synthase Deficiency
Zainab Almasseri1, Manal Nicolas- Jilwan2, Ahmad Khaled Almadani1, Mohammad Al-Owain1,5, Rousseau Gama3,4, Raashda Ainuddin Sulaiman1,5*
1Department of Medical Genetics, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
2Department of Radiology, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
3Blood sciences, The Royal Wolverhampton NHS trust, Wolverhampton, UK
4School of Medicine and Clinical Practice, Wolverhampton University, UK
5College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
Abstract
6-Pyruvoyl-tetrahydropterin synthase (PTPS) deficiency results in depletion of the brain neuro-transmitters serotonin and dopamine. Since dopamine is the physiological inhibitor of pituitary prolactin secretion, hyperprolactinemia is common in patients with PTPS deficiency. Serum prolactin concentrations are used for the monitoring and optimization of L-Dopa therapy. We report three adult patients with PTPS deficiency who had persistent hyperprolactinemia unresponsive to high dose L-Dopa therapy, and pituitary imaging confirmed microadenoma. In the presence of prolactinoma, serum prolactin is an unreliable tool for treatment monitoring in these patients. This report emphasizes the need to exclude other causes of hyperprolactinemia including prolactinoma, in patients who are compliant with optimized L-Dopa treatment and their prolactin levels remain significantly high.
Introduction
6-Pyruvoyl-tetrahydropterin synthase (PTPS) deficiency (MIM: 261640), an autosomal recessive inherited metabolic disorder, is the most common cause of tetrahydrobiopterin (BH4) deficient hyperphenylalaninemia. PTPS has an important role in the synthesis of BH4, which is an essential cofactor for a group of enzymes including phenylalanine hydroxylase, tyrosine and tryptophan hydroxylases. BH4 deficiency results in impaired hydroxylation of phenylalanine to tyrosine, tyrosine into L-Dopa and tryptophan into 5-hydroxytryptophan. PTPS deficiency, therefore, leads to hyperphenylalaninemia and depletion of the brain neurotransmitters serotonin and dopamine1. This disorder is relatively common in Saudi Arabia due to a founder tribal mutation2,3. It mostly presents in infancy with developmental delay, seizures, and abnormal movements associated with hyperphenylalaninemia. The diagnosis is made by abnormal urinary pterin metabolites (low biopterin and high neopterin), reduced levels of cerebrospinal fluid (CSF) neurotransmitters homovanillic acid (HVA) and 5-hydroxy indoleacetic acid (5-HIAA) and the presence of a pathogenic mutation in the PTS gene. Management of PTPS deficiency includes administration of synthetic BH4, and neurotransmitter replacement therapy with L-3,4-dihydroxyphenylalanine (L-Dopa) and 5- hydroxytryptophan. Clinical monitoring of this treatment may be compromised by the side-effects of L-Dopa and 5-hydroxytryptophan, a presentation which can also mimic the symptoms of their deficiency and therefore treatment is monitored by serum prolactin and CSF neurotransmitters concentrations.
Dopamine is the major physiological inhibitor of pituitary prolactin secretion and its deficiency results in mild to moderate hyperprolactinemia in patients with PTPS deficiency. It is secreted by the hypothalamus, and delivered to anterior pituitary via hypothalamohypophyseal blood circulation. Dopamine exerts its inhibitory role by binding to D2 and D4 receptors in the pituitary lactotrophs leading to down-regulation of prolactin gene expression, reduced prolactin secretion, and decreased lactotroph proliferation4,5. Serum prolactin concentrations are inversely related to concentrations of CSF- HVA (a dopamine metabolite) and are, therefore, used as a biochemical marker for optimization of L-Dopa therapy in these patients6-8. Here we report three adult patients with PTPS deficiency with persistent marked hyperprolactinemia unresponsive to high dose L-Dopa therapy, and subsequent pituitary gland MRI revealed microadenoma.
Case 1
A 31-year-old female was born preterm at 32 weeks of gestation. Her birth weight was 1.75 kg. She had a normal post-natal course. At the age of 4 months, she was noted to have hypotonia which led to the diagnosis of PTPS deficiency based on high plasma phenylalanine levels, its response to BH4 loading test, low biopterin with high neopterin levels in urine and subsequently confirmed by genetic analysis. Following diagnosis, she was treated with BH4 supplement, L-Dopa (Sinemet) and 5-hydroxytryptophan. In childhood, she developed seizures and acute dystonia which were treated with sodium valproate and adjustment of neurotransmitter dosage respectively. She has intellectual disability, mild residual dystonia and abnormal behavior with poor social and communication skills. She had primary amenorrhea and associated osteoporosis treated with alendronate. The amenorrhea due to hypogonadotrophic hypogonadism was likely secondary to persistent hyperprolactinemia. Her serum prolactin concentrations remained high despite high dose L-Dopa (10.5 mg/kg body weight/day). Adequate dopamine replacement was consistent with satisfactory CSF neurotransmitter concentrations (Table 1). MRI scan of pituitary gland showed a 3 mm microadenoma (Figure 1a). At the age of 29 year, she was referred to the endocrinologist and was started on cabergoline 0.5 mg twice weekly. Serum prolactin levels normalized and menstrual periods began. Cabergoline was stopped after six months and she continues to have menstruation. Serum prolactin concentrations, however, are gradually rising, the latest being 308 µg/L. A repeat pituitary gland MRI after a two year interval re-demonstrated unchanged microadenoma.
Table 1: Clinical and biochemical characteristics of patients
|
|
Case 1 |
Case 2 |
Case 3 |
|
Gender |
Female |
Female |
Female |
|
Age at diagnosis |
4 months |
1 month |
3 months |
|
Initial presentation |
Hypotonia |
Asymptomatic |
Hypotonia |
|
Mutation in PTS gene |
c.166_168del:p.56_56del |
c.166_168del:p.56_56del |
c.242A>G:p.E81A |
|
Primary amenorrhea |
Present |
Present |
Present |
|
Galactorrhea |
Absent |
Absent |
Absent |
|
Serum Prolactin (ref. 3.4-24µg/L) |
350-730 |
300-568 |
296-500 |
|
Serum FSH (IU/L) |
<0.1 |
1.1 |
5 |
|
Serum LH (IU/L) |
<0.1 |
1.5 |
3.3 |
|
Serum estrogen (pmol/L) |
94 |
69 |
46 |
|
Serum TSH (ref. mU/L) |
2.1 |
0.83 |
2.2 |
|
CSF studies (on treatment) 5HIAA (ref. 67-140 nmol/L) |
263 |
220 |
- |
|
HVA (ref. 145-324 nmol/L) |
649 |
549 |
- |
|
Phenylalanine (ref.13-17µmol/L |
13 |
12 |
- |
FSH: follicle stimulating hormone; LH: leutinising hormone; TSH: thyroid stimulating hormone; HVA: homovanillic acid; 5HIAA: 5-hydroxyindolacetic acid
Case 2
This 29-year-old female is the younger sister of case 1. She was born by caesarian section because of decreased fetal movement at 34 weeks of gestation. Her birth weight was 2.1 kg. PTPS deficiency was diagnosed on a newborn screen with hyperphenylalaninemia and a typical urine biopterin-neopterin profile. She was started on BH4 supplement, L-Dopa and 5-hydroxytryptophan in the neonatal period. She has a mild intellectual disability. She was noted to have hyperprolactinemia therefore, the dose of L-Dopa was gradually increased to 13 mg/kg body weight daily. However, serum prolactin concentration remained very high, the likely cause of primary amenorrhea due to hypogonadotropic hypogonadism (Table 1). Pituitary gland MRI showed a 5 mm size microadenoma (Figure 1b). She was seen by the endocrinologist and received cabergoline 0.5mg twice per week for eight months. Prolactin concentrations normalized (6.4 µg/L), and menstrual periods started. She has been started on alendronate as her bone densitometry scan showed osteoporosis.
Case 3
A 26-year-old female was diagnosed as having PTPS deficiency at the age of three months and was started on replacement therapy. She has a mild intellectual disability, severe attention deficit hyperactive disorder and behavioral problems. She was noted to have high prolactin (296 µg/L) at the age of 16 years. MRI scan of pituitary gland did not show any focal lesion. Following increment in the L-Dopa dosage, prolactin concentration initially reduced to 47.8 µg/L, then rose to 257 µg/L in a few months. She was seen by the gynecologist at age 20 years, for primary amenorrhea. Serum prolactin levels were high with secondary hypogonadotropic hypogonadism (Table 1). She was started on cabergoline 0.5 mg twice weekly, later increasing to 1 mg twice weekly. Serum prolactin levels fell (6 µg/L), and she started menstruating. Cabergoline was stopped after ten months and was treated with hormone replacement therapy (HRT; progyluton) for bone protection and regular menstruation. A year later, she was commenced on escitalopram because of psychiatric problems. Subsequent prolactin concentration after six months was 475 µg/L, in the presence of regular menstruation. MRI scan of the pituitary gland showed 3 mm microadenoma (Figure 1c, d). She was again prescribed cabergoline for 12 weeks by the gynecologist and prolactin levels came down to 89 µg/L. She was seen by the endocrinologist and was treated with HRT alone as she continued to have regular menstrual periods. Her latest prolactin on HRT is 161 µg/L. Repeat pituitary gland MRI after three years demonstrated an unchanged microadenoma.
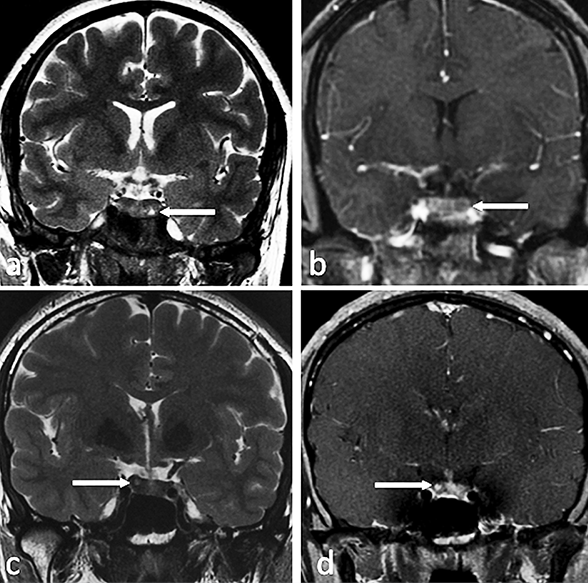
Figure 1: Magnetic resonance imaging of pituitary gland
a: Coronal T2-weighted image in patient 1 shows a small T2 hyperintense lesion in the left inferior aspect of the pituitary gland suggestive of a microadenoma (arrow), b: coronal dynamic postcontrast T1-weighted image in patient 2 shows a small rounded hypoenhancing lesion in the left aspect of the pituitary gland compatible with a microadenoma, c: coronal T2-weighted image in patient 3 shows a small T2 hyperintense lesion in the right side of the pituitary gland compatible with a microadenoma (arrow). d: coronal postcontrast T1-weighted image in patient 3 demonstrates the corresponding hyperenhancement in relation to the remainder of the gland (arrow).
Discussion
Hyperprolactinemia due to dopamine deficiency is common in patients with PTPS deficiency. Serum prolactin is used as an index of dopaminergic homeostasis and is an effective tool for monitoring and optimizing the dopaminergic replacement in PTPS deficiency. Patients are treated with L-Dopa in divided doses varying between 7-12 mg/kg body weight daily. Serum prolactin concentrations are measured three hours after the administration of L-Dopa to assess dopaminergic activity and therapeutic control.
Our adult cohort of 27 patients with PTPS deficiency consists of 16 females and 11 males. Hyperprolactinemia was observed in 10 females (range:45-100 µg/L in six and >200 µg/L in four females) and three males (range: 40-100 µg/L). Prolactin levels normalized in all male and four female patients following optimization of L-Dopa replacement. In the remaining six female patients, only the three patients described here had, persistent marked hyperprolactinemia which resulted in secondary hypo-gonadotrophic hypogonadism, manifesting as primary amenorrhea, delayed poor development of secondary sexual characters and osteoporosis. All were compliant with high dose L- Dopa replacement and in the two patients in whom CSF examination was undertaken, it confirmed adequate replacement with neurotransmitters (Table 1). Pituitary MRI demonstrated the presence of microadenoma in all patients. Since other causes of hyperprolactinemia including macroprolactinemia (Table 2) were excluded, we suggest that these patients had concurrent microprolactinomas.
Table 2: Causes of hyperprolactinemia
|
Physiological
Non-pathological |
Pregnancy, lactation, nipple stimulation, stress, exercise
Macroprolactinemia
|
|
Pharmacological |
Antipsychotics- Phenothiazines, haloperidol, risperidone, clozapine, olanzapine |
|
|
Antidepressants- Tricyclics monoamine oxidase inhibitors, selective serotonin reuptake inhibitors |
|
|
Antihypertensives – Verapamil, reserpine, methyldopa |
|
|
Antiemetics - Metoclopramide, domperidone, |
|
|
H2 blockers: Cimetidine, ranitidine |
|
Pathological (Hypothalamic and pituitary lesions) |
Prolactinoma, non-functioning pituitary adenoma causing stalk effect |
|
|
Lymphocytic hypophysitis |
|
|
Craniopharyngioma, Meningioma, Germinoma, empty sella syndrome |
|
|
Inï¬ltrative diseases – Sarcoidosis, Histiocytosis X, Tuberculosis, Metastasis |
|
Other diseases |
Primary hypothyroidism, chronic renal failure liver cirrhosis, ectopic secretion of prolactin Chest-wall lesions (trauma, surgery, herpes zoster virus), polycystic ovarian syndrome, seizures |
Prolactinomas are the most common pituitary adenomas which account for 50-60% of all pituitary tumors9. They are largely microprolactinomas (less than 1 cm in size) which are much common in females. Microprolactinomas may remain silent or cause symptoms related to hyperprolactinemia such as oligo/amenorrhea, galactorrhea and infertility in premenopausal women. Diagnosis of prolactinoma is based on the presence of hyperprolactinemia with radiological evidence of pituitary adenoma. The degree of hyperprolactinemia generally correlates with prolactinoma size. Prolactin concentration is usually100 to 250 µg/L in microprolactinomas9 but such values are not absolute10 and may range from 32 to 525 µg/L11.
Prolactinoma has not been reported before in patients with PTPS deficiency. The diagnosis of prolactinoma in these patients could be delayed by the overlap of concomitant hyperprolactinemia seen in partially L-Dopa supplemented patients. If patient is compliant with optimal L-Dopa dosage and prolactin levels remain significantly high, we suggest it is important to exclude other causes of hyperprolactinemia including concurrent prolactinoma by pituitary imaging. In these patients, serum prolactin will be an unreliable indicator of L-Dopa replacement.
They had no other features to suggest that prolactinoma was related to genetic syndromes such as multiple endocrine neoplasia type 1(MEN1) or familial isolated pituitary adenoma (FIPA). The prolactin concentrations in these three patients were higher than expected for the size of pituitary adenoma. The presence of microadenoma in these adequately replaced patients is, however, significant and consistent with a microprolactinoma as it is well recognized that L-Dopa fails to suppress serum prolactin in prolactinomas12. Treatment with cabergoline normalised serum prolactin and initiated the menstrual cycle, and following withdrawal of cabergoline therapy the hyperprolactinemia has recurred. Since the restoration of fertility was not the goal of treatment in these adults, further treatment with cabergoline was not deemed necessary at this stage.
In summary, persistent hyperprolactinemia despite adequate L-Dopa replacement should prompt investigation for other causes of hyperprolactinemia including prolactinoma. Serum prolactin may be an unreliable marker of adequacy of L-Dopa replacement therapy in co-incidental microprolactinomas.
Future studies are required to better understand the precise mechanisms of action of dopamine, dopamine receptors and their effect on prolactin gene expression in the pathophysiology of the development of prolactinoma in these patients with disproportionately high prolactin concentrations as compared to the size of adenoma, especially in the background of adequate replacement with dopaminergic neurotransmitters.
References
- Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis. 2009; 32(3): 333-342.
- al Aqeel A, Ozand PT, Gascon G, et al. Biopterin-dependent hyperphenylalaninemia due to deficiency of 6-pyruvoyl tetrahydropterin synthase. Neurology. 1991; (5): 730-737.
- Almannai M, Felemban R, Saleh MA, et al. 6-Pyruvoyltetrahydropterin Synthase Deficiency: Review and Report of 28 Arab Subjects. Pediatr Neurol. 2019; 96: 40-47.
- Cabrera-Reyes EA, Limon-Morales O, Rivero-Segura NA, et al. Prolactin function and putative expression in the brain. Endocrine. 2017; 57: 199–213.
- Samperi I, Lithgow K, Karavitaki N. Hyperprolactinaemia. J Clin Med. 2019; 13: 8(12).
- Spada M, Ferraris S, Ferrero GB, et al. Monitoring treatment in tetrahydrobiopterin deficiency by serum prolactin. J Inherit Metab Dis. 1996; 19(2): 231-233.
- Birnbacher R, Scheibenreiter S, Blau N, et al. Hyperprolactinemia, a tool in treatment control of tetrahydrobiopterin deficiency: endocrine studies in an affected girl. Pediatr Res. 1998; 43(4 Pt 1): 472-477.
- Ogawa A, Kanazawa M, Takayanagi M, et al. A case of 6-pyruvoyl-tetrahydropterin synthase deficiency demonstrates a more significant correlation of L-Dopa dosage with serum prolactin levels than CSF homovanillic acid levels. Brain Dev. 2008; 30(1): 82-85.
- Wong A, Eloy JA, Couldwell WT, et al. Update on prolactinomas. Part 1: Clinical manifestations and Diagnostic Challenges. J Clin Neurosci. 2015; 22: 1562-1567.
- Casanueva FF, Molitch ME, Schlechte JA, et al. Guidelines of the Pituitary Society for the diagnosis and management of prolactinomas. Clinical Endocrinology. 2006; 65: 265–273.
- Vilar L, Freitas MC, Naves LA, et al. Diagnosis and management of hyperprolactinemia: Results of a Brazilian multicenter study with 1234 patients. J. Endocrinol. Investig. 2008; 31: 436–444.
- Blackwell RE. Diagnosis and management of prolactinomas. Fertil Steril. 1985; 43: 5-16.