
The challenges of cystinuria in the twenty-first century - a mini review
Louise F. Øbro1, Katja V. Pedersen2, Søren K. Lildal1, Susanne S. Osther1, Helene U. Jung1, Kim H. Andreassen1 and Palle J. S. Osther1,*
1Department of Urology, Urological Research Center, Lillebaelt Hospital, University of Southern Denmark, Fredericia, Denmark
2Department of Clinical Genetics, Lillebaelt Hospital, University of Southern Denmark, Vejle, Denmark
Abstract
Cystinuria is a rare genetic disorder caused by mutations in the genes that encode the two subunits of amino acid transport, resulting in failure of absorption of filtered dibasic amino acids including cystine in the proximal tubules. Despite new knowledge of the molecular basis of cystinuria, it continues to be one of the most challenging stone diseases. There is no curative treatment of cystinuria, and patients will have life-long risk of stone formation, repeated surgery, impaired renal function and quality of life. Management of cystinuria requires a multi-modal approach in dedicated centres to improve treatment outcome and patient compliance. Recent developments in cystine crystal growth inhibitors may hold promise for more effective stone prevention in the future.
Introduction
Cystinuria is a rare genetic condition that continues to be one of the most challenging kidney stone diseases. It is caused by mutations in the genes that encode the two subunits of the amino acid transport, resulting in failure of absorption of filtered dibasic amino acids including cystine in the proximal tubules1. Since cystine has a very limited solubility in the physiological range of urine pH, this leads to complicated and recurrent kidney stone formation, harbouring a higher risk of chronic kidney disease than the more common calcium oxalate kidney stone disease. Cystinuria represents approximately 1% of adult and 3–10% of pediatric stone disease1,2. The worldwide prevalence of cystinuria is approximately 1 in 7000 neonates, ranging from approximately 1:2000 at the Mediterranean East Coast to 1:100000 in Sweden2,3.
Genetic basis of cystinuria
The inheritance pattern of cystinuria is complex. Traditionally, cystinuria has been classified into three different subtypes. Later this classification was criticised, and a new classification based on the affected genes SLC3A1 and SLC7A9 was made by Della Strologo et al.4.
Cystinuria type A is caused by mutations in the SLC3A1 gene located on chromosome 2. The SLC3A1 gene encodes the heavy subunit of the renal amino acid transporter (rBAT), which is needed to localize the transporter to the plasma membrane5. The mode of inheritance appears to be truly autosomal recessive and the penetrance is high6. An affected person has two faulty copies of the gene and each of the parents carries one copy. Carriers usually have normal level of urine cystine and they have no increased risk of developing urolithiasis compared with the general population5.
Cystinuria type B is caused by mutations in the SLC7A9 gene located on chromosome 19. The SLC7A9 gene encodes the light subunit of the renal amino acid transporter, which compromises the catalytic, transporting component5. The mode of inheritance seems to be autosomal recessive or autosomal dominant with incomplete penetrance7. For homozygotes penetrance is similar to type A6.
Carriers such as parents, siblings and children often have raised urine cystine levels and stone formation is reported in 2-18% of cases4,6. In more rare cases the patient has mutations in both genes (SLC3A1+SLC7A9). This subgroup is classified as cystinuria type AB. The frequency is reported to be 1.2% - 4%4,8,9. Today 160 different mutations in the SLC3A1 gene and 116 in the SLC7A9 gene are listed in the Human Genome Mutation Database10.
So far no clinical differences (age of presentation, number of stone episodes, interventions) have been found between type A and type B4,9,11. The full implications of type A, B or AB status are not yet fully understood, although implications for prognosis, management and treatment have been suggested.
Diagnosis
Diagnosis is based on medical history, stone analysis, laboratory investigations and imaging. Cystinuria should always be suspected in case of early kidney stone debut (< 20 years) and a family history of cystinuria.
Stone analysis and laboratory investigations
A stone analysis should be performed whenever possible. Pure cystine stones are observed in the majority of cases12. Microscopy of urine revealing hexagonal crystals are pathognomonic 2,13. Cyanide-nitroprusside colorimetric test detects cystine in urine at a threshold concentration of 75 mg/l. A quantitative chromatographic analysis of a 24-hour urine-sample confirms the diagnosis. The normal rate of cystine excretion is around 0.10 mmol/day; homozygote cystinurics usually excrete more than 1.7 mmol/day.
Imaging
Cystine stones are weak radiopaque on plain radiographs. The attenuation value (Hounsfield Unit, HU) of cystine stones on Computerized Tomography (CT) varies14. A low HU may support the suspicion of cystine stones, but there is significant overlapping between stones of different compositions, especially for cystine and uric acid stones, making a definitive differentiation challenging15-17. Dual-energy CT and automated image processing may improve the differentiation18,19. CT may also be helpful in predicting fragility of the stone by evaluating morphology of the stone, inhomogeneous stones with void regions being more fragile20-22 (Figure 1). Renal ultrasonography is less accurate but represents the mainstay for follow-up of cystine stone formers to avoid frequent exposure to radiation.
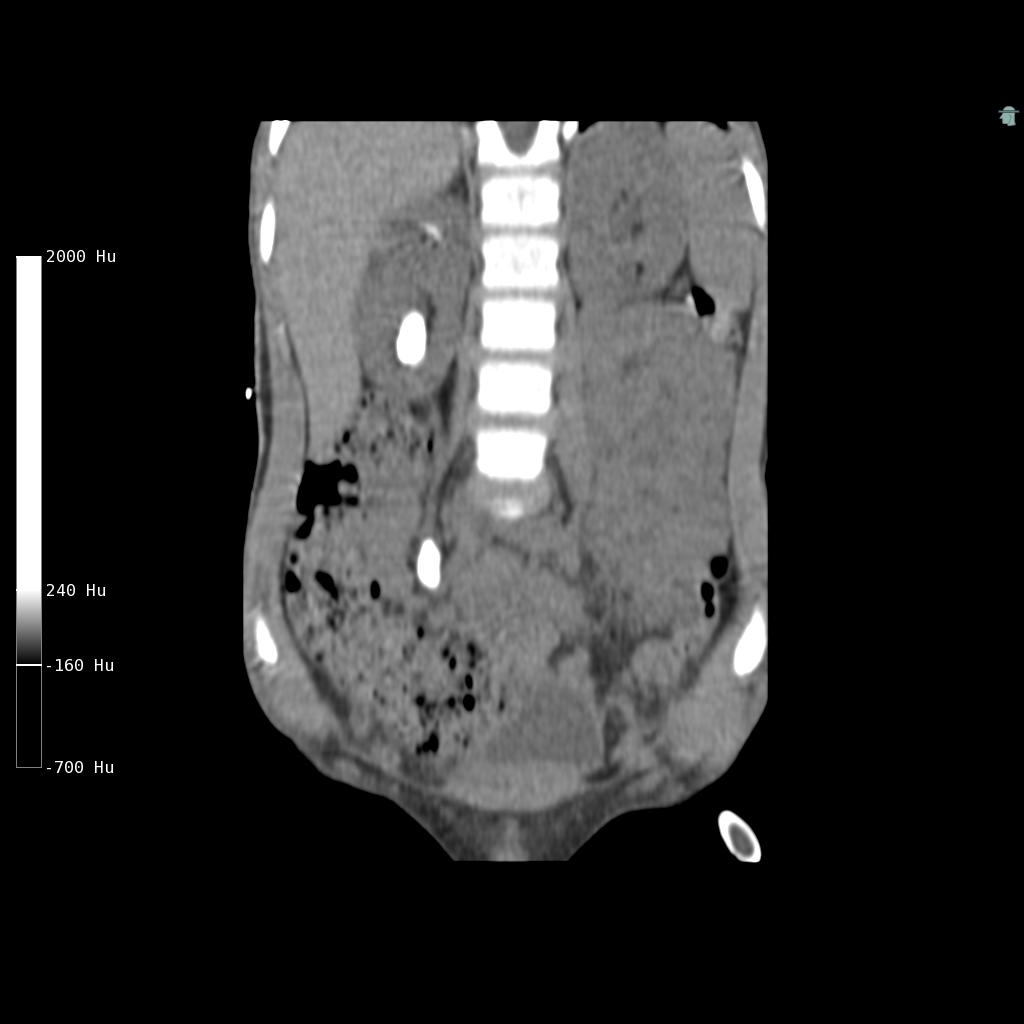
Figure 1: Low-dose non-contrast CT (coronal view) in a 6-year-old boy with cystinuria showing a large kidney stone and a very large ureteric stone on the right side. The stones are homogeneous with a Hounsfield Unit of 900, which made them inappropriate for Shock Wave Lithotripsy. The stones were treated using a percutaneous surgical approach. Cystinuric patients often presents with bilateral high stone burden disease either as staghorn stones or large rounded stones as in this case. The boy has remained stone free on a combination of hydration and potassium citrate therapy.
Surgical management
Despite aggressive medical therapy, cystinuric patients are likely to suffer frequent recurrent episodes of stones necessitating urologic intervention13. Cystinuric patients will often have experienced previous invasive procedures, demanding use of the least invasive methods to minimize complications and morbidities of multiple procedures11. On the other hand, since residual fragments after stone treatment in cystinuric patients sooner or later will result in symptomatic stone disease, complete stone clearance is of upmost importance. Thus, the surgical challenge in cystinuria is to find the balance between the safety of the procedures and the need for complete stone clearance23.
Cystine calculi are generally considered Shock Wave Lithotripsy (SWL) resistant. Pre-treatment imaging may reliably predict outcome of SWL, however; heterogeneous stones being more fragile to shock waves (SW) than homogeneous stones21. Thus, patients suitable for SWL may be selected by the appearance of the stone on Non-Contrast CT (NCCT). Smaller (<2 cm) SW resistant cystine calculi may effectively be managed ureteroscopically using flexible ureteroscopes and laser lithotripsy11,23,24, whereas larger and branched (staghorn) stones most effectively are managed by percutaneous nephrolithotomy (PCNL). Newer approaches of PCNL with minituarization of the tract (Mini PCNL) may be especially suitable for cystinuric patients reducing morbidity related to repeated renal accesses. No systemic therapies can effectively dissolute cystine stones. A significant increase in cystine solubility begins, when pH exceeds 7.025. Chemolytic dissolution of cystine stones through a nephrostomy tube or ureteral catheter may be done with the highly alkaline solution THAM (tris-hydroxymethyl-amminomethane) or the chelating solution acetylcysteine 2%, or preferably the two solutions in combination26,27. Instillation therapy for cystine stones are normally performed secondary to SWL, PCNL or ureterorenoscopy for dissolution of residual stone fragments, since chemolysis monotherapy of most cystine stones will require long periods of irrigation25,27. Due to advances in minimal invasive procedures chemolytic procedures are only rarely needed11.
Stone prevention
The mainstay of treatment consists of urinary dilution, urinary alkalinization and reduction of cystine to its more soluble metabolite, cysteine, using thiol-binding agents28,29.
There are no controlled prospective trials using stone formation as an endpoint for any therapy for cystinuria. However, studies using endpoints of cystine excretion and studies of stone passage using patients as their own historical controls provide reasonable data on which to base therapeutic choices.
Dietary measures
Urinary dilution: Increasing urine flow rate will reduce cystine concentration, thereby decreasing level of cystine saturation in urine. The level of urine flow required is dependent on the patient’s cystine excretion, but most adult patients need to produce at least 3.5 liters of urine per day to prevent stone formation. It is usually recommended that the patient awaken at least once per night to void and drink additional water28,29.
Sodium: It has been shown in several studies that sodium loading increases urinary cystine excretion, and that sodium restriction is anticystinuric30-32. Cystinuric patients should be advised to reduce sodium intake (< 2 g/day)27. Dietary intake of sodium may be assessed by urine sodium excretion.
Protein: Potentially, protein restriction should be attractive in cystinuric patients in order to reduce intake of the sulphur-containing amino acid methionine, from which sulphur is derived for cystine formation. Only one study has explored the quantitative effect of protein restriction on cystine excretion33. In this study a low-protein diet reduced cystine excretion by 20% compared to a high-protein diet. A reduction of that magnitude seems to be very modest in light of the potential malnutrition problems a low-protein diet might induce in this population. On the other hand, it seems advisable to maintain a normal protein intake, and avoid protein gluttony, since this additionally will reduce the dietary acid load, thereby increasing urine pH and cystine solubility34,35. The dietary content of protein should not exceed 0.8 g/kg body mass/day.
Pharmacologic treatment
Urinary alkalinisation: Cystine solubility increases with increasing pH, however, solubility does not increase significantly until a level of urine pH above 7-7.5 is reached. To achieve this goal requires significant and frequent doses of alkali, what the majority of patients find difficult to comply30. Furthermore, the optimal dose of alkali is difficult to estimate in any given patient, since it has been shown that cystine solubility varies considerably between individuals36. Urine dilution in itself results in an increase in pH, and the combination of urine dilution and alkali therapy is the fundamental first step in cystinuria management. Furthermore, increasing urine pH increases the cystine-solubilizing efficacy of thiol drugs (see below)37. Potassium alkali seems to be preferable, since sodium alkali will increase cystine and calcium excretion, the latter potentially increasing the risk of calcium phosphate crystallization in an alkaline urine28, 29, 35. Patient-monitored control of urine pH is important for successful alkalinisation therapy.
Thiol-binding agents: Sulfhydroxyl compounds, such as D-penicillamine or tiopronin (α-mercaptopropionylglycine), can reduce the formation of cystine crystals by reacting with cystine and generating more soluble mixed disulfide compounds (drug-cysteine complex). No RCTs are available, but both drugs seem to have the capability of reducing stone recurrences38-41. Both D-penicillamine and tiopronin have significant side effects within the usual range of therapeutic dosage (1000-2000 mg/day), including foul odor, nausea, fever, fatigue, skin rash, premature skin aging, proteinuria and hypersensitivity42, which restricts their use to patients who are unable to control stone formation with high fluid intake, dietary modification and urine alkalinisation (Figure 2). Side effects of thiols seem to be less prominent with the use of tiopronin compared to D-penicillamine39. If the patients are intolerant to D-penicillamine and α-mercaptopropionylglycine, the thiol captopril may be tried, although the role of captopril in the treatment of cystinuria is debatable43,44. In vitro findings have shown that increasing urine pH greatly increases the effect of thiol drugs to solubilize cystine in a clinically relevant time frame, highlighting the importance of combining thiol with alkali therapy37. Investigation of newer thiol-containing drugs (cysteamine and 2,3 dimercaptosuccinic acid (DMSA)) has not yet lead to clinical use45,46.
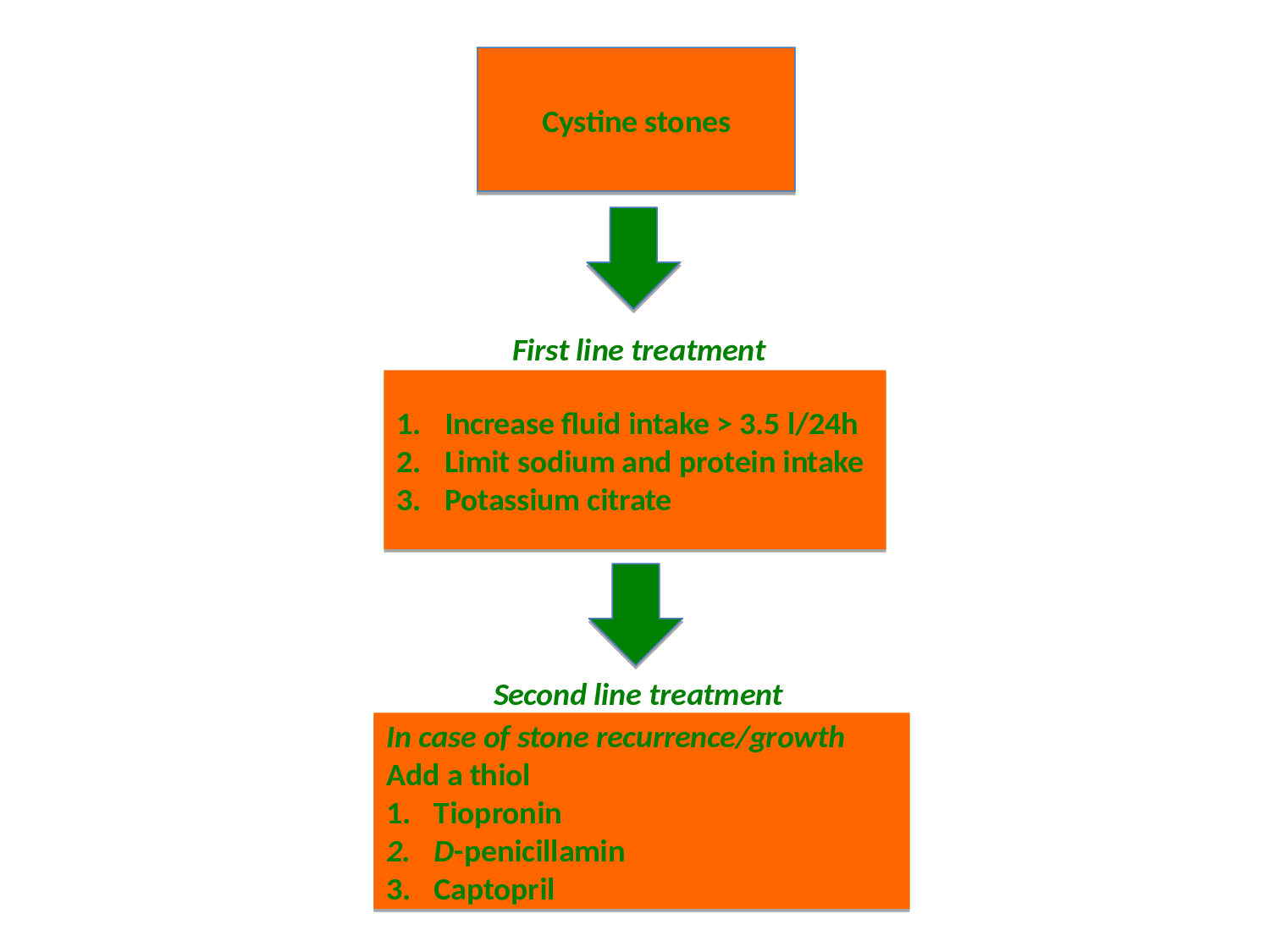
Figure 2: Algorithm for stone prevention in cystinuric patients
Cystinuria treatment pipeline
Despite above mentioned treatment options, a significant proportion of patients experience severe relapsing renal stone disease, and new more effective preventive treatment options are highly warranted. Recently, the application of atomic force microscopy to cystinuria has identified L-Cystine dimethylester (CDME) as an effective molecular imposter of L-Cystine, capable of inhibiting crystal growth in vitro47. The potential of CDME was further highlighted by demonstration of its ability to inhibit cystine crystal growth in vivo utilizing a murine model48. Subsequent development of L-Cystine diamides more effective than CDME with respect to sustaining higher concentrations of L-Cystine in solution49, which is tantamount to inhibition of crystal growth, might represent a long-awaited breakthrough in cystinuria management. Future clinical trials hopefully will confirm these promising results.
Follow-up and patient compliance
Close follow-up of patients are important to identify stone recurrences and monitor side-effects of pharmacologic therapy. Cystine stones in the early course of a recurrence tend to be heterogeneous, highlighting the importance of regular follow-up (every 3-6 months), in order to offer the patients the least invasive treatment options.
Failure of pharmacological therapy may to a large extend be contributed compliance problems. It has been shown that the need for urological intervention for recurrent stones decreased with medical compliance, and that introduction of a dedicated cystinuria clinic halved intervention rate50. Centralizing the management of patients with cystinuria to dedicated centers, therefore, may be an equally important step towards better management of this complicated group of stone patients. An algoritm for the medical management of cystinuria is presented in Figure 2.
Conclusion
Evaluating and treating cystinuria continues to represent a major challenge. Pharmacotherapy of this serious stone disease has not improved in decades despite new knowledge on the molecular-genetic basis of the disease, however, recent studies implying cystine growth inhibition through molecular mimicry, may introduce a completely new paradigm for effective stone prevention in cystinuria. Cystinuric patients should be managed in dedicated centres offering minimal invasive treatment modalities, enabling personalized treatment and increasing patient compliance.
References
- Chillaron J, Font-Llitjos M, Fort J, et al. Pathophysiology and treatment of cystinuria. Nat Rev Nephrol. 2010; 6:424–434.
- Knoll T, Zöllner A, Wendt-Nordahl G, et al. Cystinuria in childhood and adolescence: recommendations for diagnosis, treatment, and follow-up. Pediatr Nephrol. 2005; 20:19–24.
- Krombach P, G W-N, Knoll T. Cystinuria and Cystine Stones. In: Rao PN, Kavanagh JP, Preminger GM (eds) Urinary Tract Stone Disease. Springer-Verlag, London; 2011.
- Dello Strologo L, Pras E, Pontesilli C, et al. Comparison between SLC3A1 and SLC7A9 cystinuria patients and carriers: a need for a new classification. J Am Soc Nephrol. 2002; 13:2547–2553.
- Eggermann T, Venghaus A, Zerres K. Cystinuria: an inborn cause of urolithiasis. Orphanet J Rare Dis. 2012; 7:19
- Eggermann T, Zerres K, Nunes V, et al. Clinical utility gene card for: Cystinuria. Eur J Hum Genet. 2012; 20(2).
- Barbosa M, Lopes A, Mota C, et al. Clinical, biochemical and molecular characterization of cystinuria in a cohort of 12 patients. Clin Genet. 2012; 81:47–55.
- Font-Llitjós M, Jiménez-Vidal M, Bisceglia L, et al. New insights into cystinuria: 40 new mutations, genotype-phenotype correlation, and digenic inheritance causing partial phenotype. J Med Genet. 2005; 42:58–68.
- Wong KA, Mein R, Wass M, et al. The Genetic Diversity of Cystinuria in a UK Population of Patients. BJU Int. 2015 Jul; 116(1):109-16.
- Human Genome Mutation Database [Internet]. April 2015. Available from: http://www.hgmd.cf.ac.uk.
- Thomas K, Wong K, Withington J, et al. Cystinuria - a urologist’s perspective. Nat Rev Urol. 2014; 11:270–277.
- Tiselius HG. New horizons in the management of patients with cystinuria. Curr Opin Urol. 2010; 20:169–173.
- Saravakos P, Kokkinou V, Giannatos E. Cystinuria: current diagnosis and management. Urology. 2014; 83:693–699.
- Patel SR, Wagner LE, Lubner MG, et al. Radiopacity and hounsfield attenuation of cystine urolithiasis: case series and review of the literature. J Endourol. 2014; 28:472–475.
- Torricelli FC, Marchini GS, De S, et al. Predicting urinary stone composition based on single-energy noncontrast computed tomography: the challenge of cystine. Urology. 2014; 83:1258–1263.
- Motley G, Dalrymple N, Keesling C, et al. Hounsfield unit density in the determination of urinary stone composition. Urology. 2001; 58:170–173.
- Mattoo A, Goldfarb DS. Cystinuria. Semin Nephrol. 2008; 28:181–191.
- Ferrandino MN, Pierre SA, Simmons WN, et al. Dual-energy computed tomography with advanced postimage acquisition data processing: improved determination of urinary stone composition. J Endourol. 2010; 24:347–354
- Chevreau G, Troccaz J, Conort P, et al. Estimation of urinary stone composition by automated processing of CT images. Urol Res. 2009; 37:241–245.
- Bhatta KM, Prien EL, Dretler SP. Cystine calculi--rough and smooth: a new clinical distinction. J Urol. 1989; 142:937–940.
- Kim SC, Hatt EK, Lingeman JE, et al. Cystine: helical computerized tomography characterization of rough and smooth calculi in vitro. J Urol. 2005; 174:1468–70; discussion 1470.
- Kim SC, Burns EK, Lingeman JE, et al. Cystine calculi: correlation of CT-visible structure, CT number, and stone morphology with fragmentation by shock wave lithotripsy. Urol Res. 2007; 35:319–324.
- Biyani C, Cartledge JJ. Cystinuria - Diagnosis and management. EAU-EBU Updates Series. 2006; 4:175–183.
- Azili MN, Ozcan F, Tiryaki T. Retrograde intrarenale surgery for the treatment of renal stones in children: Factors influencing stone clearance and complications. J Pediatr Surg. 2014; 49: 1161-5.
- Elkoushy MA, Violette PD, Andonian S. Percutaneous instillation of chemolytic, chemotherapeutic, and antifungal agents. In: Smith AD, Badlani GH, Preminger GM, Kavoussi LR. Smith’s Textbook of Endourology. 3rd edition. Blackwell Publishing Ltd.; 2012. pp 290-309.
- Hesse A, Tiselius H-G, Siener R, et al. Cystine stones. In: Urinary stones. Diagnosis, treatment, and prevention of recurrence. 3rd edition. Basel: S. Karger AG; 2009. pp 124-141.
- Ng CS, Streem SB. Contemporary management of cystinuria. J Endourol. 1999; 13(9): 647-51.
- Xu H, Zisman AL, Coe FL, et al. Kidney stones: an update on current pharmacological management and future directions. Expert Opin Pharmacother. 2013; 14:435–447.
- Tracy CR, Pearle MS. Update on the medical management of stone disease. Curr Opin Urol. 2009; 19(2): 200-4.
- Goldfarb DS, Coe FL, Asplin JR. Urinary cystine excretion and capacity in patients with cystinuria. Kidney Int. 2006; 69:1041-47.
- Peces R, Sanchez L, Gorostidi M, et al. Effects of variation in sodium intake on cystinuria. Nephron. 1991; 57:421- 423.
- Rodriguez LM, Santos F, Malaga S, et al. Effect of a low sodium diet on urinary elimination of cystine in cystinuric children. Nephron. 1995; 71:416-8.
- Rodman JS, Blackburn P, Williams JJ, et al. The effect of dietary protein on cystine excretion in patients with cystinuria. Clin Nephrol. 1984; 22:273-278.
- Rogers A, Kalakish S, Desai RA, et al. Management of cystinuria. Urol Clin North Am. 2007; 34(3): 347-62.
- Sumorok N, Goldfarb DS. Update on cystinuria. Curr Opin Nephrol Hypertens. 2013; 22(4): 427-31.
- Nakagawa Y, Asplin JR, Goldfarb DS, et al. Clinical use of cystine supersaturation measurements. J Urol. 2000; 164:1481-1485.
- Asplin DM, Asplin JR. The interaction of thiol drugs and urine pH in the treatment of cystinuria. J Urol. 2013; 189(6): 2147-51.
- Dolin DJ, Asplin JR, Flagel L, et al. Effect of cystine-binding thiol drugs on urinary cystine capacity in patients with cystinuria. J Endourol. 2005; 19:429-432.
- Harbar JA, Cusworth DC, Lawes LC, et al. Comparison of 2-mercaptopropionylglycine and D-penicillamine in the treatment of cystinuria. J Urol. 1986; 136:146-9.
- Barbey F, Joly D, Rieu P, et al. [Medical treatment of cystinuria: evaluation of long-term results in 30 patients]. Presse Med. 2000; 29:528-32.
- Tekin A, Tekgul S, Atsu N, et al. Cystine calculi in children: the results of a metabolic evaluation and response to medical therapy. J Urol. 2001;165: 2328-30.
- Dello Strologo L, Laurenzi C, Legato A, et al. Cystinuria in children and young adults: success of monitoring free-cystine urine levels. Pediatr Nephrol. 2007; 22: 1869-73.
- Cohen TD, Streem SB, Hall P. Clinical effect of captopril on the formation and growth of cystine calculi. J Urol. 1995; 154:164-166.
- Perazella MA, Buller GK. Successful treatment of cystinuria with captopril. Am J Kidney Dis. 1993; 21:504-7.
- Belldina EB, Huang MY, Schneider JA, et al. Steady- state pharmacokinetics and pharmacodynamics of cysteamine bitartrate in paediatric nephropathic cystinosis patients. Br J Clin Pharmacol. 2003; 56:520-525.
- Parvex P, Rozen R, Dziarmaga A, et al. Studies of urinary cystine precipitation in vitro: ontogeny of cystine nephrolithiasis and identification of meso-2,3-dimer- captosuccinic acid as a potential therapy for cystinuria. Mol Genet Metab 2003; 80: 419-425.
- Rimer JD, An Z, Zhu Z, et al. Crystal growth inhibitors for the prevention of L-cystine kidney stones through molecular design. Science. 2010; 330:337-341.
- Lee MH, Sahota A, Ward MD, et al. Cystine growth inhibition through molecular mimicry: a new paradigm for the prevention of crystal diseases. Curr Rheumatol Rep. 2015;17:33.
- Hu L, Yang Y, Aloysius H, et al. l-Cystine Diamides as l-Cystine Crystallization Inhibitors for Cystinuria. J Med Chem. 2016; 59:7293-7298.
- Haritopoulus K, Fojtik P, Cross W, et al. Impact of a metabolic stone clinic on management of patients with cystinuria: 5 years follow-up. Clin Ter. 2010; 161: 342-344.