
Ayami Sato1,2, Nantiga Virgona2, Yuko Sekine1 and Tomohiro Yano2*
1Graduate School of Medical and Pharmaceutical Sciences, Chiba University, Chiba, Japan
2Research Institute of Life Innovation, Toyo University, Gunma, Japan
Malignant mesothelioma (MM) is an aggressive cancer associated with exposure to asbestos. In recent years, despite restrictions on the use of asbestos, the incidence of MM has been increasing due to its long latency period. Owing to its poor prognosis, the treatment of MM requires innovative therapies. Tocotrienol (T3), one of the vitamin E analogues, has powerful antioxidant properties and anti-cancer effects. However, these effects have not been fully understood, and are mediated independent of its antioxidant activity. Therefore, we have synthesized a new redox-inactive derivative of T3 (T3E) that has shown a stronger anti-MM effect than its redox-sensitive mother compound. In this review, we discuss the potential for anti-MM effect of T3E as non-antioxidant functions of T3 by introducing our previous reports.
Malignant mesothelioma (MM) is a particularly aggressive cancer which originates in the mesothelial cells lining the body cavities, such as the pleural cavity. MM is mainly attributed to asbestos exposure. Although the use of asbestos has already been restricted in many countries, the incidence of MM has continued to rise owing to its long latency period, of around 40 years1. Currently, a combination of chemotherapy (cisplatin/pemetrexed), surgery, and radiotherapy provides the best long-term results. However, even after such an aggressive approach the prognosis remains poor, with mean patient survival time of just over one year2. This is primarily due to the resistance of MM to treatment, and therefore new therapeutic approaches to MM are urgently required. It is suggested that several signal molecules related to growth and survival are constitutively activated in MM cells3. Accordingly, simultaneous suppression of these multi-target molecules by therapeutic agents could offer a novel approach to MM treatment.
Vitamin E is composed of two structurally similar compounds, tocopherols (T) and tocotrienols (T3), both made up of a chromanol ring with an isoprenoid-derived hydrophobic tail, (Figure 1a). The presence of a phenolic hydroxyl (-OH) group on the chromanol ring is a necessity for the antioxidant activity of vitamin E. However, the differences in side chains between T3 and T contributes to the different potentials of various functions including anti-cancer effects4,5. Unlike T, T3 posess an unsaturated isoprenoid side chain that endows superior anti-cancer activities4. Vitamin E compounds have been extensively used in pharmacological studies due to their anti-oxidant properties; however, a major difference exists between T and T3. It has been shown that T3 is both a potent down regulator of HMG-CoA reductase, the rate-limiting enzyme of the mevalonate pathway for cholesterol biosynthesis, and an inhibitor of cancer cell proliferation6. The pro-apoptotic effect of the vitamin E family and its analogues on cancer cells is mediated independently of their antioxidant properties and that the synthetic derivatives of the naturally occurring subset of these compounds may improve this effect7. For example, a synthetic redox-inactive derivative of T bearing a succinyl ester at the phenolic -OH group of the chromane ring, α-tocopheryl succinate (TS) is one such candidate, (Figure 1b). It has been established that TS exhibits greater apoptotic induction in cancer cell lines than its parent compound8, as well as a cytotoxic effect in various malignant tumors including MM9. However, the cytotoxic effect of TS has been abrogated in cells and tissues that easily hydrolyze the ester bond of TS and convert it back to T10. Thus, it is more useful to block the antioxidant property of vitamin E by use of a nonhydrolyzable ether bond11. Recently, T3 has received greater research interest and has been shown that it is a superior anti-cancer agent to TS12. Unfortunately T3 has short elimination half life in vivo, due to high metabolic rate and antioxidant property13,14.To overcome this disadvantage of T3 usage in vivo, it seems that a redox-inactive analogue of T3 including the ether bond may act as a potential anti-cancer agent comparing with that of T such as TS. Based on this speculation, we synthesized a new redox-inactive derivative of T3, 6-O-carboxypropyl-α-T3 (T3E) by reacting 4-hydroxybutanenitrile with 4-bromobutyronitrile, (Figure 1b), to reinforce the anti-cancer effect and improve and confirmed reinforcement of T3E compared with T3 in human cancer cells15.
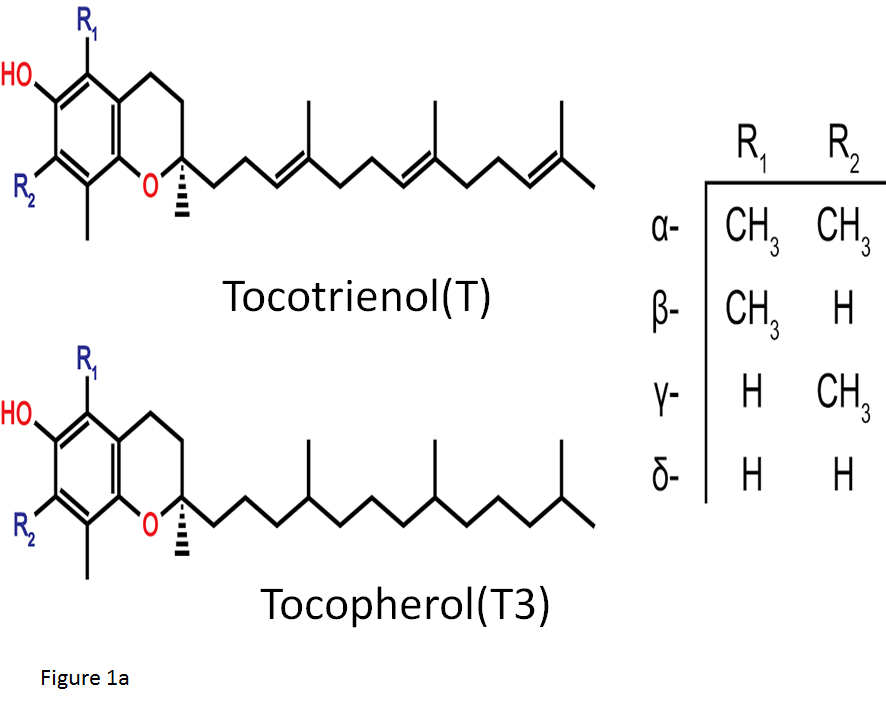
Figure 1a: Natural analogues of Vitamin E
The structure of the two natural groups of Vitamin E: tocopherols and tocotrienols. The -OH group located on the chromane ring conveys the redox action of these molecules whilst the side chains contrast the two classes in terms of non-redox activities.
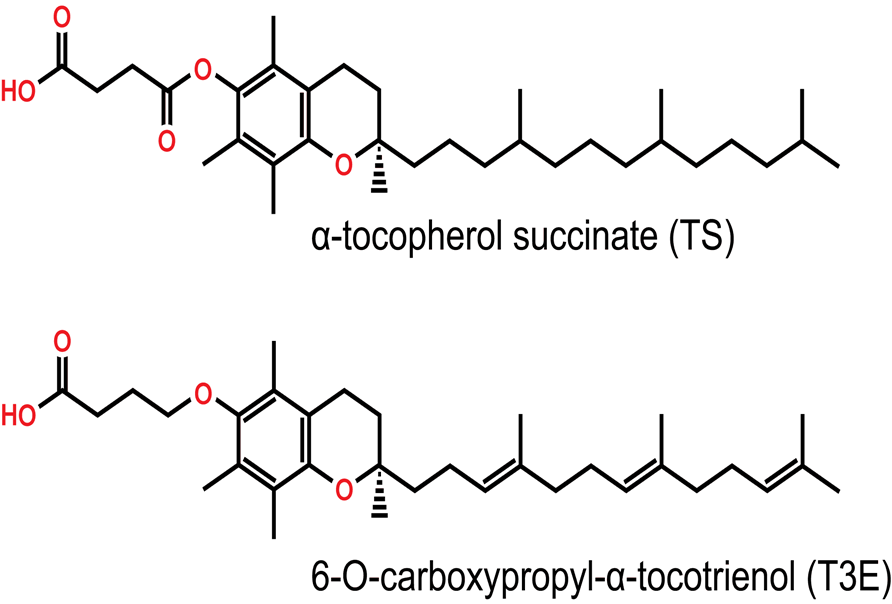
Figure 1b: Synthetic derivatives of Vitamin E
The structure of the redox-inactive derivatives of the α forms of tocopherol and tocotrienol. Inactivation occurs at the phenolic -OH group through either esterification in α-tocopherol succinate (TS) or etherification in 6-O-carboxypropyl- α-tocotrienol (T3E).
A previous study has shown that anti-cancer agents having weak acids are selectively taken up by malignant cells due to the acidic interstitium of the tumors16, so the addition of chargeable group on the chromanol ring of T3 may contribute to the selectivity of T3E against the malignant tumor cells. We have confirmed T3E accumulation into cancer cells and a longer elimination half life in vivo when compared with T3 (unpublished data). Overall, it seems to be possible that T3E especially induces strong cytotoxicity on MM cells compared with other cancer cells, due to the difference of microenvironments. Thus, in this mini-review, we focus on the anti-MM effect of T3E and try and clarify the possible mechanism of T3E-induced cytotoxic effect in MM cells, (Figure 2).
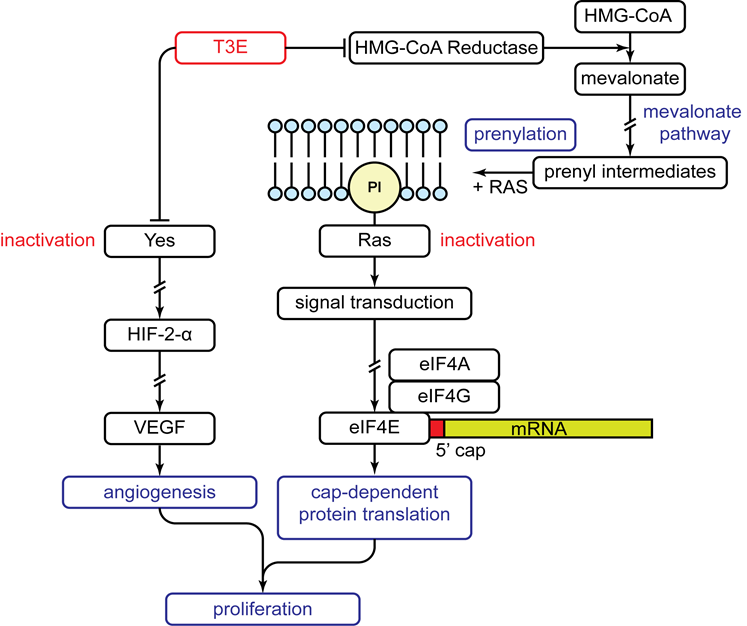
Figure 2: Anti-proliferative effect of T3E on human malignant mesothelioma cells
Mechanism of the anti-proliferative effects of T3E on human malignant mesothelioma cells as discovered through our research. T3E suppresses both the activity and expression of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase. The result is inactivation of Ras as it is unable to be prenylated causing an inability to activate cap-dependent protein translation through eukaryotic initiation factor 4E (eIF4E). Simultaneously, T3E directly inhibits Yes causing down regulation of downstream targets: hypoxia inducible factor-2α (HIF-2α) and vascular endothelial growth factor (VEGF). The overall effect is a suppression of angiogenesis and proliferation. Arrows and perpendicular lines indicate activation and inhibition respectively while broken lines indicate omitted steps.
Our previous studies have shown that T3E could effectively suppress the growth of human MM cells having chemo-resistance to cisplatin in pharmacological doses (10-20μM) that were treated for 48 h. In addition, the cytotoxic effect of T3E on MM cells was much stronger than that of T3 with a lesser toxic effect on non-tumorigenic mesothelial cells (Met5A cell line)17. Thus, we used the pharmacological doses in all of the reported experiments.
Inhibition of the mevalonate pathway results in reduced production of prenyl intermediates. These molecules are required in the activation of Ras through prenylation. Ras, a proto-oncogene, is a critical component in intracellular signal transduction pathways. Thus, Ras inactivation would interrupt cell signalling causing an arrest of cell proliferation and because of this, it is an inviting target for the design of chemotherapeutic agents18. Statins, a class of lipid-lowering drugs, that act through the inhibition of HMG-CoA reductase, resulting in Ras inactivation, have demonstrated anti-cancer effects against multiple cancers including MM19. One of our recent studies has demonstrated that treatment with atorvastatin, a member of the statin family, reduces RAS activity and MM cell viability20. Similarly, we have observed that T3E suppresses the expression of HMG-CoA reductase and subsequently inactivates RAS in MM cells5. Particularly, RAS signaling relates to aberrant cap-dependent protein translation, a critical process for the malignancy of MM20,21. Cap-dependent protein translation regulates de novo synthesis of proteins, while aberrant activation of the translation is a hallmark of many cancer types, including MM21. Protein translation is mediated by the binding of eukaryotic initiation factor 4E (eIF4E), a trimeric protein complex that binds to the 5’-cap structure (m7GpppN) of mRNA in mammalian cells. The cap-binding component of eIF4F (eIF4E) has limited availability within the cell causing it to be the rate-limiiting step of the formation of the eIF4F complex22. The activation of eIF4E is closely associated with growth regulatory factors and anti-apoptotic proteins in cancer cells. eIF4E-binding protein 1 (4E-BP1) negatively regulates eIF4E functions by blocking the interaction between eIF4E and the scaffolding protein eIF4G, thereby inhibiting the formation of the active eIF4E complex and suppressing cap-dependent protein translation22.
These suggest that inhibition of eIF4E activity leads to severe cytotoxicity via reduction in translation of specific mRNAs. Actually, we have found that T3E induces RAS inactivation and inhibits the eIF4E/eIF4G complex formation, which are associated with T3E cytotoxicity in MM cells20. Moreover, 4EGI-1, a specific inhibitor of the eIF4E/eIF4G binding, induced cytotoxicity in MM cells with reduction in the levels of ornithine decarboxylase (ODC), a growth-regulatory protein, and B-cell CLL/lymphoma 2 (BCL2), a main anti-apoptotic protein22. T3E had a similar effect on these proteins20. Overall, T3E exhibits cytotoxic effect in MM cells via disruption of the activated cap-dependent translation complex through RAS inactivation.
In addition to RAS, Src family kinases (SFKs), the intracellular non-receptor tyrosine kinases, are known as representative proto-oncogenes and candidate molecules that could hold promise in the treatment of cancer patients23. Some SFK family members have been established as malignant factors and contribute to the malignancy in MM23. Of the SFKs, Src, Yes, Fyn and Lyn show ubiquitous expression in MM cell lines23-25. In our study, it has been shown that some MM cell lines overexpressed Yes, compared with other SFK members such as c-Src and that only the silencing of Yes but not other SFK members by small interfering RNA contributes to the suppression of MM cell growth25. From the data, we have concluded that Yes has a central role inducing the appearance of malignancy in MM cells. Similarly, Yes inhibition in colon carcinoma also resulted in increased apoptosis and reduced tumor growth both in vivo and in vitro26. In solid tumors, expansion of the tumor mass is a consequence of uncontrolled cell proliferation, a hallmark of malignancy. As oxygen supply to the growing tumor becomes inadequate a hypoxic condition develops resulting in hypoxic regions of cells within the tumor27. The cells can adapt to their hypoxic condition, a process that is mainly regulated by hypoxia inducible transcription factors (HIF1 and HIF2). These HIFs coordinate the cells response in adapting to hypoxia by promoting gene expression involved in metabolism, angiogenesis, cell profliferation, differentiation and apoptosis27. The expression of HIF-2α closely relates to malignancy under hypoxia in some cancers including MM28,29. Under hypoxia, the activation of SFKs partly stabilizes HIF30, and we confirm that the level of HIF-2α is maintained by hypoxia-induced Yes activation28. Additionally, vascular endothelial growth factor (VEGF), a key molecule in the downstream pathway of Yes signaling, was secreted through HIF-2α activation under hypoxic conditions in MM cells28. In the tumors growing under hypoxia, HIF-2α induced production of VEGF, which binds to and activates VEGF receptor, thereby activating the VEGF pathway, that is central in the pathophysiology of angiogenesis, leading to adequate hypoxia adaptation of the tumor cells28. In fact, high expression of VEGF in MM is a marker of poor-prognosis31.
Additionally, our previous study has demonstrated that the survival and invasion capacity of lung cancer cells (A549 cell line) under hypoxia was suppressed by T3E via the inactivation of Src and subsequent reduction of HIF-2α level32. Taken together, T3E remarkably inhibited HIF-2α accumulation and VEGF secretion by inactivation of hypoxia-induced Yes activation. These data suggest that Yes/HIF-2α/VEGF could be new candidates for targeted treatment in MM and that T3E acts as a potent anti-MM agent.
In addition to our reports, we have determined some factors contributing to T3E-mediated anti-MM effects (unpublished data). These reports strongly support T3’s non-antioxidant functions and its use as an effective anti-cancer agent. Taken together, a redox-inactive derivative of T3 has multiple target points in MM cells, and the development of an MM treatment approach based on the non-antioxidant property of T3 is reasonable and promising.
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI; Grant-in-Aid for JSPS Fellows (15J05098).