
Update on the fluorometric measurement of enzymatic activities for Lysosomal Storage Disorder detection: The example of MPS VI.
Paula G. Franco1,2, Ana M. Adamo1,2, Patricia Mathieu1,2, María J. Pérez1,2, Patricia C. Setton-Avruj1,2 and Lucas Silvestroff1,2*
1Universidad de Buenos Aires, Facultad de Farmacia y Bioquímica, Cátedra de Química Biológica Patológica. Buenos Aires, Argentina
2Universidad de Buenos Aires, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Instituto de Química y FisicoquímicaBiológicas (IQUIFIB) Profesor Alejandro C. Paladini, Facultad de Farmacia y Bioquímica, Argentina
Abstract
Lysosomal Storage Disorders (LSD) are rare diseases that as a whole have a combined incidence ranging from 1:1500 to 1:7000 live births. One of such diseases is Mucopolysaccharidosis VI (MPS VI), or Maroteaux Lamy Syndrome. MPS VI patients undergo devastating and irreversible skeletal alterations and multisystemic failure as from early childhood due to reduced Arylsulfatse B (ARSB) enzyme activity.
Reaching a final diagnosis is not always a short cut path, but rather a years-long battle against uncertainty and unnecessary medical interventions. Our aim is to contribute from the bench table with different approaches that could serve as alternatives to pre-existing assays for screening and diagnosing MPS VI and other LSD.
The present work is based on our research article authored by Franco et al.1 where we studied the effect of blood-derived hemoglobin, and other blood components, on the fluorescence of 4-Methylumbelliferone when measuring ARSB enzyme activity from dried blood spot (DBS) samples.
Our experience indicates that to date there are plenty of different approaches for measuring ARSB enzyme activity, although the sample type required or the assay in itself often make them more adaptable for either high throughput screening or small scale diagnostics.
As a whole, the fluorometric determinations seem to be the most accessible to low budget laboratories with equally valuable performances as a sophisticated mass spectrometry analysis for this disease. Furthermore, the DBS serves as an attractive sample type for screening the disease in large populations.
Background
Mucopolysaccharidosis type VI (MPS VI), or Maroteaux Lamy Syndrome (MIM 253200), is a Lysosomal Storage Disorder (LSD) biochemically characterized by a deficiency in the lysosomal enzyme Arylsulfatase B (ARSB, EC:3.1.6.12). The consequent intracellular accumulation of the glycosaminoglycans (GAG) Dermatan Sulfate (DS) and Chondroitin Sulfate (CS) in this disease triggers multiple organ failure2. MPS VI therapy involves symptomatic treatment as well as hematopoietic stem cell transplantation and/or enzyme replacement with the human recombinant ARSB Galsulfase3. The earlier the treatment is initiated, a better clinical outcome is expected for the patient4-9. Together with other low incidence Inborn Errors of Metabolism (IEM), LSD are currently recognized as Rare Diseases and have been somewhat overlooked by the scientific community and pharmaceutical industries. As the NIH Genetics Home Reference indicates, the exact incidence of MPS VI is unknown. Its incidence varies considerably from 1:248,000 to 1:1,300,00010, with an expected global incidence of 1:340,000. As a result of this epidemiological data, research and development for better understanding the disease, and improving its diagnosis, treatment and follow up has lagged compared to other more frequent pathologies. We believe that efforts towards optimizing diagnosis and/or early screening will have an impact on the outcome of treatment.
When MPS VI is suspected, as well as for other LSD, the first approach is to confirm abnormal urinary GAG profiles10. The final diagnosis for ARSB deficiency is done by measuring the ARSB enzyme activity (EA) in dried blood spot (DBS) samples, cultured fibroblasts or blood leukocytes and/or by genetic testing10,11.
One of the most popular methods for screening and/or diagnosing MPS VI, or other LSD, uses specific 4-methylumbelliferone (β-MU)-derived synthetic substrates to measure the ARSB EA, where the hydrolysis of the substrate releases fluorescent β-MU12. The resulting fluorescence is interpolated in β-MU calibration curves for EA determination.
In our previous research article Improving arylsulfatase activity determination in dried blood spots: Screening and diagnostic approaches for Maroteaux–Lamy syndrome (MPS VI)1, we adapted the protocol of Civallero et al.13. to demonstrate the significant effect that certain DBS-derived compounds had on the β-MU fluorescence when measuring ARSB EA. We therefore provide strategies to overcome these problems both for confirmatory diagnostic tests, as well as for implementing this fluorometric method for MPS VI screening, such as a newborn screening (NBS) program. Taking into account that enzyme replacement therapy is available for the LSD Fabry Disease, Gaucher Disease, Pompe Disease, and Mucopolysaccharidosis type I, II, and VI, it is mandatory to achieve an early diagnosis and treatment initiation that could minimise the clinical severity of these diseases14,15.
Current ARSB enzymatic activity assays
Originally, ARSB was measured using the colorimetric and synthetic substrate p-Nitrocatechol Sulphate16, but its low analytical sensitivity soon became a drawback. Immunocapture methods have also been proposed17, and served both for measuring the enzyme concentration as well as its EA.
The β-MU-based substrate method has had good acceptance over the years, mainly for its economic and practical feasibility, and due to its high analytical sensitivity and specificity. The standard method uses as substrates specific residues linked to β-MU, that render the β-MU fluorescent product when the substrate is cleaved by the enzyme. Kumar et al.18 developed a novel fluorimetric EA determination for ARSB from DBS samples and using natural enzyme substrates attached to β-MU, that allow a better discrimination between EA in samples belonging to control and MPS VI patients. However, the main drawback of this method is that these new substrates are not commercially available at the moment. With this approach, Kumar et al. claimed that the specificity of their semi-natural substrate facilitated the measurement of ARSB activity without the interference of ARSA enzyme present in DBS sample18. However, others have reported that the contribution of ARSA to the EA readings was negligible when using synthetic substrates, as demonstrated by Tan et al.19 who showed that ARSA in DBS was relatively unstable. Moreover, Ba2+or Ag+ ions in the reaction buffer have been used to inhibit ARSA residual activity16,20.
Mass spectrometry is the gold standard biochemical method for diagnosing this disease21. However, not all laboratories count on the necessary equipment for developing this technique and added that it is time consuming. The high analytical and diagnostic sensitivities of this method in some cases favours numerous false positives and have raised awareness and the proposal of using second-tier tests to confirm the positive results obtained from sample analysis by MS22. Furthermore, unlike for other diseases that are diagnosed using a mass spectrometer to directly measure distinctive metabolites in biological samples, DBS samples are not run directly when looking for MPS VI cases. Instead, they are subjected to an enzymatic reaction prior to the mass spectrometry stage where only enzymatic reaction products are analyzed. However, the unbeatable advantage of this method is the possibility of performing multiplex analysis in a single run for each sample and of analyzing a very wide range of different biological compounds23,24.
The standard fluorometric determination with β-MU-derived substrates is still a robust method for measuring ARSB EA in DBS. As for other LSD EA determinations using blood-derived samples, this fluorometric method has its caveats.
Samples used for fluorometric determinations of EA
When analyzing EA using β-MU-derived substrates, testing the sample´s properties is critical for informing accurate and reliable absolute EA values. The β-MU is particularly sensitive to blood component quenching, and is mainly attributed to hemoglobin. We showed in Franco et al.1 that ARSB EA values were significantly underestimated when β-MU calibration curves were prepared in matrices different from that of the sample reaction matrix. Oemardien et al.25
proved that ridding hemoglobin from DBS-derived reaction solutions increased β-MU fluorescence for several lysosomal enzymes, however the need of a precipitation step in the protocol is a hassle.
Since DBS samples are a composite of whole blood cellular and acellular fractions, we compared the EA in other blood-derived dried samples blotted onto Guthrie card filter paper. Serum and plasma samples lacking hemoglobin do not have a significant quenching effect on β-MU fluorescence. However we found ARSB EA was lower in these samples than that measured in DBS, and may be explained by the lack of white blood cells containing lysosomes in these fractions. Other samples frequently used for measuring lysosomal EA with low/null quenching effects on β-MU fluorescence are fibroblasts or white blood cells26, although they are not easily available and are reserved for diagnostic purposes. EA in leucocytes is sensitive to long term shipping, or unfavourable transport conditions. Cultured fibroblasts require an invasive skin biopsy sample, trained personnel and time for the culture to develop27,28. We prefer DBS as the sample of choice for our protocol, since it is readily available for NBS programs and the Guthrie cards have the advantage of easy and inexpensive transport. Similar to other protocols, the determination of ARSB EA in DBS described in Franco et al.1 was adapted to use a single dried blood-impregnated filter paper disc measuring 3 mm in diameter for each sample.
In regards to the advantage of DBS, Choy et al.29 analyzed a cohort of MPS VI patients to understand under what circumstances they had reached a final diagnosis. They found that almost 30% of patients had been belatedly diagnosed due to the lack of access to, or distance from, a diagnostic facility. In this sense, the DBS stands out as a possible solution for easily transporting biological samples and thereby accelerating the diagnosis and treatment implementation. Furthermore, the DBS sample has the additional advantage of being useful for molecular testing as well30,31.
Adapting a technique for Diagnostics or Screening
Several LDS are currently been screened as part of pilot NBS programs in different countries23,32. We stand by these initiatives, and have a strong motivation driving our research in regards to LSD and the expansion of NBS programs. We figured that the technical approach to the ARSB EA measurement we put forward in Franco et al.1 could be adapted both for MPS VI diagnostics as well as for a NBS approach. At either end of the spectra, the number of patient samples analyzed at a time sets the bar in terms of costs, practicality, diagnostic specificity and sensitivity requirements.
For example, the immunocapture assay proposed by Tan et al.19 and Hein et al.17 are specific and robust methods for measuring ARSA and ARSB protein concentration and EA, but their implementation costs for high throughput screening suggest a limitation. Alternatively, Ullal et al.33 adapted a useful microplate assay for measuring ARSB EA in DBS. Although this method has a desirable format for large sample number analysis, the calibration curve they used does not seem to consider the quenching effects that blood components have on β-MU fluorescence and should be reviewed.
In Franco et al.1 we paid special attention to both sample and calibration curve matrices, and made sure the calibration curve contained DBS components as well. Moreover, we built tailor made calibration curves for each patient DBS sample. Our first contribution, was the SuSiQ Method, where the use of an internal standard provided the means for reaching true EA values and avoiding skewed results due to the differences in the sample and calibration matrices (Figure 1). The main benefit of this approach is its versatility, since it can be implemented for measuring other EA in DBS using β-MU-derived substrates.
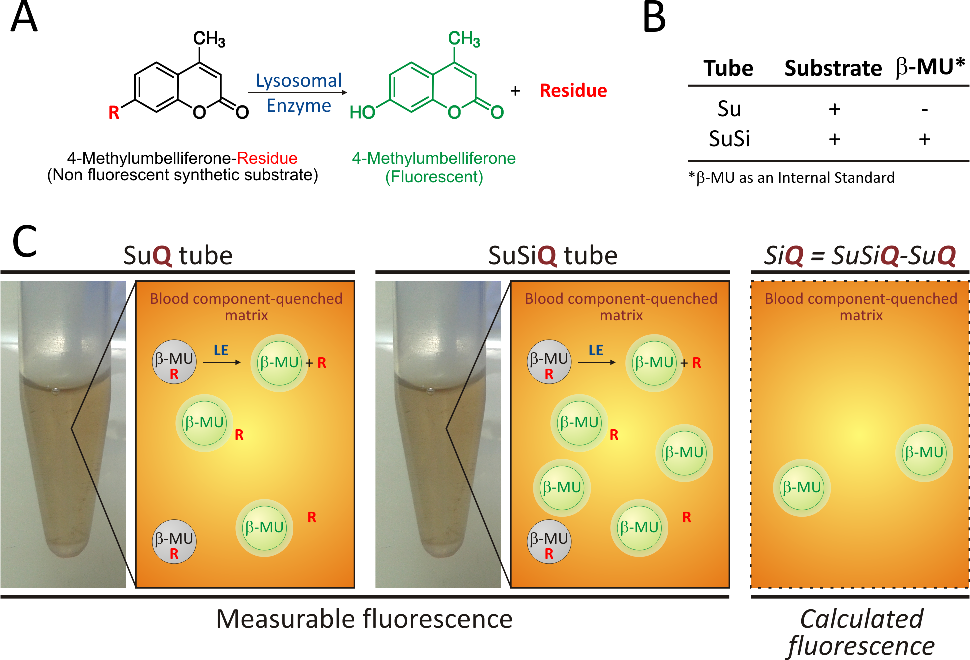
Figure 1: The SuSiQ method..
A) Example of an in vitro reaction used to measure a lysosomal enzyme activity where a synthetic β-MU-derived substrate is hydrolyzed by a specific lysosomal enzyme (LE) to give fluorescent β-MU plus a LE-specific residue (R) as products.
B) Components included in either (i) Substrate (Su) tubes or (ii) Substrate + β-MU Internal Standard (SuSi) tubes as described in Franco et al.1
C) Schematic representation of the SuSiQ method depicting how the Internal Standard fluorescence is calculated in the quenched matrix (SiQ) from data obtained from measuring the β-MU fluorescence in the SuQ and SuSiQ reaction tubes. The letter Q is used to indicate the quenching effect blood components have on β-MU fluorescence.
For the particular case of ARSB EA determinations in DBS a precipitate forms during the reaction process and appears to depend on the ARSB synthetic substrate. Therefore, for this case the use of the same DBS components in the β-MU calibration curve as in the reaction tube (as proposed for all other LSD EA determinations where no precipitate forms) is misleading. The method optimization described in Franco et al.1 for ARSB activity measurement reduced the population dispersion of EA values.
Our proposed method was only analyzed with DBS belonging to healthy individuals. The lack of access to samples of LSD patients made it impossible to calculate the assay's diagnostic sensitivity. However, the work of Ceci et al.34 using unquenched calibration curves obtained results with a diagnostic sensitivity and specificity of 100% when using DBS from Maroteaux Lamy patients. Even though their assay does not stand on its own as a final diagnostics tool, it is able to accurately differentiate healthy individuals from MPS VI affected patients with a flawless positive predictive value.
Since the blood absorbance parameter in the reaction buffer for each sample correlated with the sample quenching effect on β-MU fluorescence, we put forward a NBS approach for ARSB EA termed the Fq Method, where the absorbance was used to correct and estimate the fluorescence. This method is proposed as a first tier test that could be performed in high numbers of samples at a time, that would be necessarily followed-up by a second diagnostic test.
This is in line with the early work of Chamoles et al.35 through to that of Cobos et al.30 that suggest DBS can be used for diagnosing different MPS diseases as well.
As mentioned by Civallero et al.13 and many others, the fluorometric determination using β-MU-derived substrates can be extended to other lysosomal enzymes other than ARSB. In fact, our protocol can be equally adapted to other LSD EA determinations if the adequate synthetic substrate is used. For example, we adapted the protocol of Civallero et al.13 for measuring Total Hexosaminidase activity in DBS using the SuSiq Method and detected appreciable differences with the protocols. A few example of other lysosomal enzymes that can be measured with this protocol are alpha-galactosidase A, alpha-1,4-glucosidase, acid beta-glucosidase or alpha-L-iduronidase that are affected in patients with Fabry disease, Pompe disease, Gaucher disease or MPS I, respectively.
Concluding remarks
We think that research and development of screening and/or diagnostic tools for IEM contribute to the healthcare system as they offer alternative implementation benefits for clinical laboratories lacking access to a tandem mass spectrometer. Furthermore, the ever more frequent questioning whether LSD should be included or not in NBS programs sets a promising new stage for debate36. Ombrone et al.22 review examples of NBS programs or pilot studies that already include a handful of LSD in their first-tier tests. Matern et al.37 provide further comparative statistical data of NBS for Gaucher disease, MPS I and II, Krabbe disease, Niemann Pick A/B disease, Fabry disease, Pompe disease in Taiwan, Austria, Hungary and the US Washington State, where either fluorometric or MS techniques were used to perform the screening.
With the optimization of the analytical techniques that allow more accurate results, we hope to impact and improve the early diagnosis of MPS VI and other LSD. Notwithstanding, due to the pathology´s low incidence, the lack of biological samples from MPS VI patients in Argentina or neighbouring countries is still needed to validate our method. From the standpoint of our particular healthcare system and public funding allocation agencies, we have reached a turning point where our clinical research value would significantly improved by having access to biological samples of MPS VI or other LSD, and that this sample availability is partly hampered due to a lack of patient information centralization and epidemiological statistics. And the latter could be achieved if these diseases were included in a NBS program, for example, which takes us back to the aim of our research.
Acknowledgment
The following are institutions that support our scientific work: Universidad de Buenos Aires (UBA), the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and the Ministerio de Ciencia, Tecnología e Innovación Productiva. The authors declare they hold no conflict of interest with the funding institutions regarding the publication of this work.
References
- Franco PG, Pérez MJ, Aranda C, et al. Improving arylsulfatase activity determination in dried blood spots Screening and diagnostic approaches for Maroteaux-Lamy syndrome (MPS VI). Clin Chim Acta. 2015; 15; 446: 86-92.
- Valayannopoulos V, Nicely H, Harmatz P, et al. Orphanet J Rare Dis. 2010; 12(5): 5.
- Harmatz PR, Garcia P, Guffon N, et al. Galsulfase (Naglazyme®) therapy in infants with mucopolysaccharidosis VI. J Inherit Metab Dis. 2014; 37(2): 277-87.
- McGill J, Inwood A, Coman D, et al. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age a sibling control study. Clinical Genetics. 2010; 77: 492–498.
- Furujo M, Kubo T, Kosuga M, et al. Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI. Mol Genet Metab. 2011; 104(4): 597-602.
- Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014; 111: 63-72.
- Horovitz DD, Magalhães TS, Acosta A, et al. Enzyme replacement therapy with galsulfase in 34 children younger than five years of age with MPS VI. Mol Genet Metab. 2013; 109: 62-69.
- Pineda M, O’Callaghan M, Fernandez Lopez A, et al. Clinical Evolution After Enzyme Replacement Therapy in Twins with the Severe Form of Maroteaux–Lamy Syndrome. JIMD Reports. 2016; 30: 7-14.
- Vairo F, Federhen A, Baldo G, et al. Diagnostic and treatment strategies in mucopolysaccharidosis VI. Appl Clin Genet. 2015; 308: 245-55.
- Swiedler SJ, Beck M, Bajbouj M, et al. Threshold effect of urinary glycosaminoglycans and the walk test as indicators of disease progression in a survey of subjects with Mucopolysaccharidosis VI Maroteaux-Lamy syndrome. Am J Med Genet A 2005; 134A(2): 144-50.
- Wood T, Bodamer OA, Burin MG, et al. Expert recommendations for the laboratory diagnosis of MPS VI. Mol Genet Metab. 2012; 106(1): 73-82.
- Castilhos CD, Mezzalira J, Goldim MP, et al. Determination of the lysosomal hydrolase activity in blood collected on filter paper an alternative to screen high risk populations. Gene. 2014; 25536(2): 344-7.
- Civallero G, Michelin K, de Mari J, et al. Twelve different enzyme assays on dried-blood filter paper samples for detection of patients with selected inherited lysosomal storage diseases. Clin Chim Acta. 2006; 372(1-2): 98-102.
- Brady RO. Enzyme replacement for lysosomal diseases. Annu Rev Med. 2006; 57: 283-96.
- Kelly JM, Bradbury A, Martin DR, et al. Emerging therapies for neuropathic lysosomal storage disorders. Prog Neurobiol. 2016; S0301-0082(15): 30078-2.
- Baum H, Dodgson KS, Spencer B. The assay of arylsulphatases A and B in human urine. Clin Chim Acta. 1959(4): 453–455.
- Hein LK, Meikle PJ, Dean CJ, et al. Development of an assay for the detection of mucopolysaccharidosis type VI patients using dried blood spots. Clin Chim Acta. 2005; 353: 67–74.
- Kumar AB, Spacil Z, Ghomashchi F, et al. Fluorimetric assays for N-acetylgalactosamine-6-sulfatase and arylsulfatase B based on the natural substrates for confirmation of mucopolysaccharidoses types IVA and VI. Clin Chim Acta. 2015a; 7451(Pt B): 125-8.
- Tan MA, Dean CJ, Hopwood JJ, et al. Diagnosis of metachromatic leukodystrophy by immune quantification of arylsulphatase A protein and activity in dried blood spots. Clin Chem. 2008; 54(11): 1925-7.
- Christomanou H, Sandhoff K. A sensitive fluorescence assay for the simultaneous and separate determination of arylsulphatases A and B. Clin Chim Acta. 1977; 1579(3): 527-31.
- Kumar AB, Masi S, Ghomashchi F, et al. Tandem Mass Spectrometry Has a Larger Analytical Range than Fluorescence Assays of Lysosomal Enzymes Application to Newborn Screening and Diagnosis of Mucopolysaccharidoses Types II, IVA, and VI. Clin Chem. 2015b; 61(11): 1363-71.
- Ombrone D, Giocaliere E, Forni G, et al. Expanded newborn screening by mass spectrometry New tests future perspectives. Mass Spectrom Rev. 2016; 35(1): 71-84.
- Elliott S, Buroker N, Cournoyer JJ, et al. Pilot study of newborn screening for six lysosomal storage diseases using Tandem Mass Spectrometry. Mol Genet Metab. 2016; 118(4): 304-9.
- Chennamaneni NK, Kumar AB, Barcenas M, et al. Improved reagents for newborn screening of mucopolysaccharidosis types I, II, and VI by tandem mass spectrometry. Anal Chem. 2014; 86(9): 4508-14.
- Oemardien L, Boer A, Ruijter G, et al. Hemoglobin precipitation greatly improves 4-methylumbelliferone-based diagnostic assays for lysosomal storage diseases in dried blood spots. Mol Genet Metab. 2011; 102: 44–8.
- Camelier M, De Mari J, Burin M, et al. Extended use of dried-leukocytes impregnated in filter paper samples for detection of Pompe Gaucher and Morquio A diseases. Clin Chim Acta. 2015; 15446: 218-20.
- Coelho JC, Giugliani R. Fibroblasts of skin fragments as a tool for the investigation of genetic diseases: technical recommendations. Genet Mol Biol. 2000; 23: 269-271.
- Vangipuram M, Ting D, Kim S, et al. Skin punch biopsy explant culture for derivation of primary human fibroblasts. J Vis Exp. 2013; (77): e3779.
- Choy YS, Bhattacharya K, Balasubramaniam S, et al. Identifying the need for a multidisciplinary approach for early recognition of mucopolysaccharidosis VI (MPS VI). Mol Genet Metab. 2015; 115(1): 41-7.
- Cobos PN, Steglich C, Santer R, et al. Dried blood spots allow targeted screening to diagnose mucopolysaccharidosis and mucolipidosis. JIMD Rep. 2015; 15: 123-32.
- Verma J, Thomas DC, Kasper DC, et al. Inherited Metabolic Disorders: Efficacy of Enzyme Assays on Dried Blood Spots for the Diagnosis of Lysosomal Storage Disorders. JIMD Rep. 2016.
- Matern D, Gavrilov D, Oglesbee D, et al. Newborn screening for lysosomal storage disorders. Semin Perinatol. 2015; 3: 206-16.
- Ullal AJ, Millington DS, Bali DS. Development of a fluorometric microtiter plate based enzyme assay for arylsulfatase B (MPS VI) using dried blood spots. Mol Genet Metab Rep. 2014; 1: 465–467.
- Ceci R, de Francesco PN, Mucci JM, et al. Reliability of enzyme assays in dried blood spots for diagnosis of 4 lysosomal storage disorders. Advances in Biological Chemistry. 2011; 1: 58-64.
- Chamoles NA, Blanco M, Gaggioli D. Fabry disease enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta. 2001; 308(1-2): 195-6.
- Peake RW, Marsden DL, Bodamer OA, et al. Newborn Screening for Lysosomal Storage Disorders Quo Vadis. Clin Chem. 2016; 62(11): 1430-1438.
- Matern D, Gavrilov D, Oglesbee D, et al. Newborn screening for lysosomal storage disorders. Seminars in Perinatology. 2015; 39(3): 206–216.