
Vitamin D deficient rickets: Not always nutritional
Angham Nasser Al Mutair1,2,3, Yasser Ali Binafif1
1Department of Pediatrics, Endocrinology Division, King Abdulaziz Medical City -Riyadh, King Abdullah Specialist Children Hospital (KASCH).
2King Saud bin Abdulaziz University for Health Sciences.
3King Abdullah International Medical Research Center, Riyadh 11155, Kingdom of Saudi Arabia.
Abstract
Rickets in pediatrics due to vitamin D deficiency is still considering a major problem even in sunny and tropical countries. The prevalence of vitamin D deficiency and insufficiency in children between 6 and 15 years of age in the Kingdom of Saudi Arabia is 95.4%. Vitamin D requires two steps of hydroxylation to be functionally active. The first hydroxylation step occur in the liver by 25-hydroxylase encoded by CYP2R1 gene (11p15.2) to produce 25(OH)D3 and the second step of hydroxylation occur in proximal convoluted tubules in kidney by 1α-hydroxylase encoded by CYP27B1 gene (12q13.1) to produce hormonally active 1,25-dihydroxyvitamin D3 (1α,25(OH)2D3). Vitamin D-dependent rickets type 1B (VDDR1B) is a form of rickets due to mutation in CYP2R1 gene. Until today, only five mutations were found to affect CYP2R1 gene which lead to abnormal structure and function of 25-hydroxylase enzyme. Presence of symptoms of vitamin D deficiency in good dietary history with poor response or depending on regular to high dose of vitamin D2 or D3 to maintain 25(OH)D3 level should raise the suspicion of genetic causes of CYP2R1 mutations. These mutations are inherited as autosomal recessive; the severity and response to medication depend on number of allele affected. Some of the homogenous mutation patients showed some improvement in using Calcitriol. Bypassing the first step of hydroxylation (25-hydroxylase) in liver using 25-OH-D3 (Calcifediol) as treatment lead to dramatic improvement of biochemical and radiological finding in seven recent reported cases which needs further study.
Introduction
Classical vitamin D deficiency remains the major cause of rickets among children worldwide. A combination of poor nutritional, social, and climatic conditions explains much of this deficiency. Even in sunshine countries such as Saudi Arabia, rickets remains prevalent in different age groups1. The prevalence of vitamin D deficiency (25-hydroxyvitamin D3 [25(OH)D3] is ≤25 nmol/L) and vitamin D insufficiency (25(OH)D3 level is 25-50 nmol/L) in children and adolescents between 6 and 15 years of age in the Kingdom of Saudi Arabia is 45.5% and 49.9% respectively, over all 95.4%2.
An understanding of the physiology of vitamin D metabolism and its action is important to know the pathology and genetic causes of vitamin D deficiency. The source of vitamin D in the body is through the conversion of 7-dehydrocholesterol to vitamin D3 in the skin upon exposure to sunlight ultraviolet-B and through ingestion of foods containing either cholecalciferol (vitamin D3) or ergocalciferol (vitamin D2) or through supplements of these substances. Once produced in the skin or absorbed from the intestine, vitamin D (including D2 and D3) requires two steps of hydroxylation to be in active and physiologic form. The first hydroxylation step occurs in the liver by 25-hydroxylase (CYP2R1, 11p15.2) converting vitamin D3 to 25(OH)D3 and the second step of hydroxylation occur in proximal convoluted tubules in kidney by 1α-hydroxylase (CYP27B1, 12q13.1) to convert 25(OH)D3 to hormonally active 1,25-dihydroxyvitamin D3 (1α,25(OH)2D3) (figure 1). The active form of vitamin D has important roles in maintaining normal calcium and phosphate level by enhancing absorption of these elements from kidney and intestine which are important in bone mineralization. Defect in any of these steps will lead to effect on calcium and phosphate hemostasis and bone mineralization that will lead to rickets in children and osteomalacia in adults3,4.
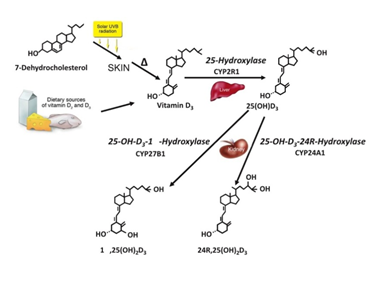
Figure 1.Metabolism of vitamin D3 in the skin, liver and kidney. 7-Dehydroxholesterol in the skin is converted in two steps and beginning with the exposure to ultraviolet light to vitamin D3, which is transported in the blood (mostly protein-bound) to the liver where the side chain is hydroxylated at the C25-position. The resulting 25(OH)D3 is the major circulating form of vitamin D. In the kidney, 25(OH)D3 is hydroxylated at either the C1- or the C24 position to form either 1a,25(OH)2D3 or 24R25(OH)2D3. The cytochrome P450 (CYP) dependent enzymes responsible for these transformations are, respectively, the 25-hydroxylase (CYP2R1), the 1a-hydroxylase (CYP27B1) and the 24R-hydroxylase (CYP24A1 or CYP24). Helen L. Henry copyright 2011. Modified with permission
Helen L. Henry. Regulation of vitamin D metabolism. Best Practice & Research Clinical Endocrinology & Metabolism, BPRCEM. 2011; 25:531–541.
Defect in 1α-hydroxylase enzyme due to mutation in CYP27B1 gene resulting in defect the synthesis of active form of vitamin D (1α,25(OH)2D3), this condition called Vitamin D-dependent rickets type 1A (VDDR1A; MIM 264700). Vitamin D-dependent rickets type 1B (VDDR1B; MIM 600081) is a form of rickets due to mutation in CYP2R1 gene encoding 25-hydroxylase. Vitamin D-dependent rickets type 2A (VDDR2A; 277440) is caused by end-organ unresponsiveness of active vitamin D due to mutation in the gene encoding the vitamin D receptor. Vitamin D-dependent rickets type 2B (VDDR2B; MIM 600785) is an unusual form of end-organ unresponsiveness to active vitamin D due to an abnormal protein that interferes with the function of the VDR5.
Hepatic 25-hydroxylase is mitochondrial and microsomal enzyme that has six cytochrome P450 enzymes, three of them found in the animals (CYP2C11 in rat, CYP2D25 in pig, and CYP2J3 in rat) and the other three found in the human (CYP3A4, CYP27A1, and CYP2R1). CYP3A4 has activity to hydroxylate D2 but not D3 while CYP27A1 has activity to hydroxylate D3 but not D26. CYP2R1 has high affinity to hydroxylate both D2 and D3 with hydroxylation activity toward D3 equal to 26-fold higher than CYP27A1, suggesting that CYP2R1 plays the major role in vitamin D hydroxylation in humans6,7.
Experimental studies
The second hydroxylation step of vitamin D is clearly to be carried out by a single enzyme, 1α-hydroxylase coded by CYP27B1. However, it is not clear if the first hydroxylation step carried out by single or multiple enzymes responsible for 25-hydroxylation. An experimental study was done by Jinge G. Zhu et al. to evaluate the physiological role of CYP2R1 in the vitamin D activation pathway. They have created Cyp2r1−/− and Cyp2r1−/−/Cyp27a1−/− Mice (double knockout mouse models). They found that both types of mice had more than 50% reduction in 25(OH)D3 level but still measurable with normal and unchanged in 1,25(OH)2D3 concentration as compared with wild-type mice8. This finding coincides with the observation of that patients who are carrying CYP2R1 mutation had an abnormally low 25(OH)D3 measurable level with a normal 1,25(OH)2D3 concentration11,12,13,16. Also 25(OH)D3 is reduced in heterozygous but the level is in between the wild-type and homozygous levels. On the other hand, the mice with Cyp27a1−/− showed two- to threefold increase in 25(OH)D3 and this finding is identical to the previous reports8,9. CYP27A1 is responsible enzyme for cholesterol metabolism and bile acid synthesis but has a minimal effect on vitamin D metabolism. Mutation in CYP27A1 leads to defect in bile acid synthesis and this condition seen in patient with cerebrotendinous xanthomatosis9,10.
These results observed by Jinge G. Zhu et al. who supported the idea of CYP2R1 as the major and most important enzyme responsible for the first step of vitamin D hydroxylation at C-25, but not exclusive as there is another enzyme contributed to this 25 hydroxylation but to a lesser extent8.
Reported cases
From Nigeria
The first mutation reported in CYP2R1 gene was announced by S.J. Casella et al. study in 1994 in Nigerian boy and his brother. Both had typical symptoms of rickets with no history of malabsorption. 25(OH)D level was low which improved on a high dose of vitamin D (4000 IU/day) and back to a low level with modest dose of vitamin D (1000 IU/ day). Genetic test done and showed the first mutation reported in CYP2R1 gene which is c.296T > C (L99P)11.
Cohort study was conducted by Tom D. Thacher et al. about the prevalence of CYP2R1 mutation in Nigerian children. They found a new novel mutation in addition to the previously known reported mutation of CYP2R1 in two out of twelve families. One mutation was c.296T > C (L99P) found in 8 members of 2 generations of both families. The other mutation was c.726A > C (K242N) found in heterozygous mother of the index case in the second family12. These mutations effect CYP2R1 structure and function. L99P amino acid replacement will effect on CYP2R1 folding while K242N amino acid change will affect interaction surface of CYP2R1 with vitamin D13,14.
The first family had L99P mutation of CYP2R1 in form of homozygous (the index case, his brother and father) and heterozygous (2 sisters and mother) with more severe rickets seen in homozygous members. In homogenous patients, index case and his brother were presented at age of 12.5 and 11 years respectively. Both were had legs deformity, bone pain, difficulty in walking, signs of rickets, and history of low calcium rich diet associated with severe biochemical and radiological picture of rickets. In heterozygous members, only one sister had a history of mild genu varum that resolved spontaneously, others were normal. The second family had heterozygous L99P mutation of CYP2R1 in index case, her sister and father. Both sisters were had typical clinical, biochemical, and radiological pictures of rickets12.
As seen in experimental and Al sagheir A. et al studies, 25(OH)D3 level was low in heterozygous patients but in between the homozygous and normal8,12,16. Homozygous patients (L99P) showed no response to the treatment with 50000 IU of vitamin D compared with heterozygous patients (L99P and K242N). They demonstrated that the incremental levels of 25(OH)D2 and 25(OH)D3 at day three (the peak) after supplementing D2 and D3 were blunted in homozygous while partial increments in heterozygous compared with control (significant P value) group. In L99P homozygous subjects, the 1,25(OH)2D was relatively low and increased with supplement of D2 and D3.12 On the other hand, the improvement in rickets clinically and radiologically were seen after using very high dose of vitamin D, 600,000 IU IM, indicating CYP2R1 mutation can be overcome by high dose of vitamin D as seen in the previous experimental study which showed low but measurable 25(OH)D3 in Cyp2r1-/- mice8,12. This observation supporting that there might be a role of other P450 enzymes in 25 hydroxylation.
From Saudi Arabia
Two new mutations in CYP2R1 were described by Al Mutair A. N. et al. in two siblings of Saudi family. The index case was 13 years old girl first presented to clinic at age of 10 years as short stature. The second sibling was the older brother 14.7 years old who had symptoms at age of 5 years. Both siblings had bow legs, bone pain, and decrease activity in addition to difficulty in walking and other signs of rickets seen in older brother. They had normal development, nutritional, antenatal and postnatal history in addition to absent of malabsorption symptoms. Both were on vitamin D supplement at different doses and the brother used additionally calcium supplement. Initial investigation showed classical biochemical picture of vitamin D deficiency in presence of normal 1,25(OH)2D3 (table 1)15.
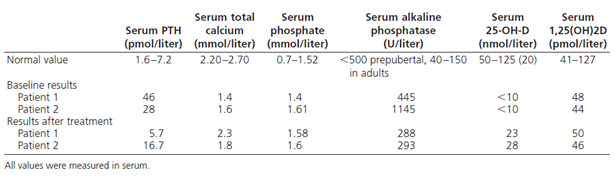
table 1.Biochemical results (baseline and after therapy)
Angham N. Al Mutair, Ghada H. Nasrat, and David W. Russell. Mutation of the CYP2R1 Vitamin D 25-Hydroxylase in a Saudi Arabian Family with Severe Vitamin D Deficiency. The Journal of Clinical Endocrinology & Metabolism, JCEM. 2012; 97(10):E2022–E2025
Both siblings were treated with Cholecalciferol (5000 IU/day for the girl and 10,000 IU/day for the boy). As seen in table 1, the abnormal biochemical results improved with treatment except 25(OH)D3 level ,which was increased but remain low in spite of high dose treatment, coincides with experimental study in mice8,15. Serum level of 1,25(OH)2D3 remained normal with treatment reflecting normal one alfa hydroxylase enzyme activity that is under parathyroid hormone control. The unaffected sibling had normal serum 25(OH)D3 level without vitamin D supplementation. Their mother had vitamin D deficiency by history and on regular supplement of vitamin D to maintain 25(OH)D3 in normal level. Father refused to be tested. These finding rose suspicion of selective 25-hydroxyvitamin D3 deficiency15.
Molecular genetic testing was done for the family and showed new not previously reported compound heterozygous mutations (figure 2) in both siblings. One mutation was inherited from the father with transition of G to A in the first nucleotide of the highly conserved splice donor sequence of intron 2 (c.367+1,G>A). This mutation will cause abnormal splice in mRNA that will cause truncated CYP2R1 protein. The second mutation was inherited from the mother with T insertion in exon 3 (c.768,insT) that alters the normal translational reading frame. This mutation will cause again truncated CYP2R1 protein. Both truncated CYP2R1 proteins lack the domains required for normal function of 25-hydroxylase enzyme. These mutations has autosomal recessive inheritance pattern (figure 2)15.
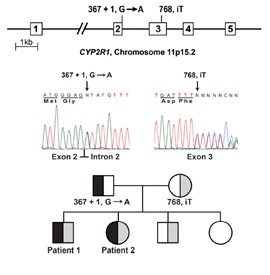
Figure 2.DNA sequence of CYP2R1 in patients 1 and 2 and pedigree of family. Affected individuals inherited one CYP2R1allele with a G to A transition in the splice donor sequence of intron 2 and a second allele with a T insertion in exon 3. The inheritance of these alleles in the family is shown.
Angham N. Al Mutair, Ghada H. Nasrat, and David W. Russell. Mutation of the CYP2R1 Vitamin D 25-Hydroxylase in a Saudi Arabian Family with Severe Vitamin D Deficiency. The Journal of Clinical Endocrinology & Metabolism, JCEM. 2012; 97(10):E2022–E2025
A second study done by Al sagheir A. et al. reported the clinical and molecular characterization of 25-Hydroxylase deficiency in 30 Saudi patients who presented with classical symptoms of vitamin D deficiency and low 25(OH)D3 (<50 mmol/L),who poorly responded to the usual treatment doses of vitamin D, and had no other medical problems including nutrition disorder16. Two mutations were found similar to the mutations reported by Al Mutair A. N. et al: c.367+1,G>A (44.4%) and c.768,insT (55.6%), 18 patients were had homozygous mutations, and heterozygous mutations were found in 9 patients from two families with rickets (familial rickets). There were no mutation detected in 3 patients who had no family history of rickets (non familial rickets)15,16.
Homozygous patients had more severe symptoms and lower 25(OH)D3 as compared to heterozygous patients, as seen in the experimental study and the previous Saudi siblings reports8,15,16. The most clinical presentations were bone pain, limitation in activity, bone deformities and hypocalcaemia with percentage 86.2%, 58.6%, 31%, and 17.2% respectively16. All heterozygous patients and most of homozygous patients (72.2%) were responded to a high dose of cholecalciferol (50,000-100,000 IU/week). The remaining of homozygous patients (27.8%) were responded to Calcitriol16.
From France
Arnaud Molin et al. discovered a new mutation in CYP2R1 that is not previously reported (c.124_138delinsCGG [p.Gly42_Leu46delinsArg]) in one family. Interestingly, a selective 25-hydroxy vitamin D therapy (Calcifediol) was tried as treatment in difficult diagnosed patients. They described 7 patients from two unrelated families, one from France and the other from Moroccan extraction. The proband case of France family was 4 years old boy, on Cholecalciferol 1000-1200 IU/day till age of 18 months. He developed leg deformity, bone pain and hypotonia. Biochemical and radiological investigation showed typical picture of vitamin D deficiency. Initially diagnosed as VDDR1A and given Alfa calcidol which lead to partial improvement of biochemical parameters except parathyroid hormone (PTH) and alkaline phosphatase (ALP) but later he developed hypercalcaemia and hypercalciuria as complications of treatment. Once diagnosed as VDDR1B with homozygous mutation, shifted to Calcifediol (25-OH-D3) which showed marked improvement in all biochemical parameters. The proband case of Moroccan family was 9 years old boy had difficulty in walking at age of 5 years. He was initially on Cholecalciferol 800 IU/day till the age of 18 months then he was put on 100,000 IU vitamin D yearly. At 7 years of age he presented with proximal muscle weakness and diagnosed as myopathy with normal muscle biopsy. Later he was found to have homozygous mutation in CYP2R1. Both patients had low serum 25(OH)D3 level and high 1,25(OH)2D3. Another 5 members from Moroccan family were had homozygous mutations in CYP2R1 with very low serum 25(OH)D3 level that normalized after using Calcifediol at different doses17. Genetic study showed a new mutation in CYP2R1 gene in family 1 (c.124_138delinsCGG [p.Gly42_Leu46delinsArg]), confirmed in vitro as a loss of function mutation17.
This study explore the difficulty in diagnosing patients with VDDR1B mutations and this is the first study used Calcifediol (25-OH-D3) as treatment; bypass the first step of hydroxylation (25-hydroxylase) in liver, which lead to dramatic improvement of biochemical and radiological finding in all 7 cases with both homozygous mutations (c.124_138delinsCGG [p.Gly42_Leu46delinsArg] and c.296T>C [p.Leu99Pro])17.
Conclusion
Rickets due to vitamin D deficiency is still considered as a major problem in pediatrics even in sunny and tropical countries.1 This problem will be more prevalent if there are multiple risk factors present together like poor nutritional, social, and climatic conditions, and it is rarely due to genetic causes. One of the genetic defects is related to 25 hydroxylase enzyme found in liver which is mainly coded by CYP2R1 gene located at chromosome 11p15.2; other genes are involved in 25-hydroxylation but to a lesser extent. CYP2R1 is responsible for normal structure and function of 25 hydroxylase enzyme3,4,6,7.
Until today, only five mutations were found to affect CYP2R1 gene. Two mutations were reported from Nigeria (c.296T > C [L99P] and c.726A > C [K242N]), another 2 were from Saudi Arabia (c.367+1,G>A and c.768,insT), and the recent one from France (c.124_138delinsCGG)11, 12,15,17.
As observed from the previous studies, symptoms of vitamin D deficiency with normal dietary history and poor response of 25(OH)D3 level to D2 or D3 treatment especially with strong family history of vitamin D deficiency should raise the suspicion of genetic causes for CYP2R1 mutations (VDDR1B) . Patients with CYP2R1 mutations have normal or high 1,25(OH)2D3 (if received vitamin D) and they need supra-physiological doses of Cholecalciferol to see full or partial improvement11,12,15,16,17.
The phenotype appearance in CYP2R1 mutations (VDDR1B) seem to be more complicated and depend on the number of allele affected as compared with autosomal recessive VDDR1A12. Patient with one allele affect has modest low of 25(OH)D3 and will respond to regular vitamin D supplement compared with two alleles affect patient with severe low of 25(OH)D3 who may respond to higher dose of cholecalciferol or Calcitriol12,15,16. Using Calcifediol (25-OH-D3) as treatment; will bypass the first step of hydroxylation (25-hydroxylase) in liver, which lead to dramatic improvement of biochemical and radiological finding in seven cases from a single report that may need further studies17.
References
- MS Al-Atawi, IA Al-Alwan, AN Al-Mutair, et al. Epidemiology of nutritional rickets in children. Saudi Journal of Kidney Diseases Transplantation, SJKDT. 2009; 20: 260–265.
- Adnan M, Al Shaikh, Bahaa Abaalkhail, et al. Prevalence of Vitamin D Deficiency and Calcium Homeostasis in Saudi Children. Journal of Clinical Research in Pediatric Endocrinology. JCRPE. 2016 Dec; 8(4): 461–467.
- Helen L, Henry. Regulation of vitamin D metabolism. Best Practice & Research Clinical Endocrinology & Metabolism. BPRCEM. 2011; 25: 531–541.
- Holick MF. Vitamin D: Evolutionary, physiological and health perspectives. Current Drug Targets journal. CDTJ. 2011; 12(1): 4–18.
- Vitamin D Hydroxylation-Deficient Rickets; Genetic Heterogeneity of Vitamin D-Dependent Rickets. Online Mendelian Inheritance in Man, OMIM. [Carol: 09/09/2016; 27/07/2017]. https://www.omim.org/entry/264700#description
- Cheng JB, Motola DL, Mangelsdorf DJ, et al. De-orphanization of cytochrome P450 2R1: a microsomal vitamin D 25-hydroxilase. Journal of Biological Chemistry, JBC. 2003; 278(39): 38084-38093.
- Shinkyo R, Sakaki T, Kamakura M, et al. Metabolism of vitamin D by human microsomal CYP2R1. Biochemical and Biophysical Research Communications. BBRC. 2004; 324: 451–457.
- Jinge G, Zhu, Justin T. Ochalek, Martin Kaufmann, Glenville Jones, and Hector F. DeLuca. CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proceedings of the National Academy of Sciences, PNAS. 2013; 110 (39): 15650–15655.
- Rosen H, Reshef A, Maeda N, et al. Markedly reduced bile acid synthesis but maintained levels of cholesterol and vitamin D metabolites in mice with disrupted sterol 27 hydroxylase gene. Journal of Biological Chemistry, JBC. 1998; 273(24): 14805–14812.
- Moghadasian MH. Cerebrotendinous xanthomatosis: Clinical course, genotypes and metabolic backgrounds. Clinical and investigative medicine, CIM. 2004; 27(1): 42–50.
- Casella SJ, Reiner BJ, Chen TC, et al. A possible genetic defect in 25-hydroxylation as a cause of rickets. Journal of Pediatrics, J. Pediatr. 1994;124 :929–932.
- Thacher TD, Fischer PR, Singh RJ, et al. Levine. CYP2R1 Mutations Impair Generation of 25-hydroxyvitamin D and Cause an Atypical Form of Vitamin D Deficiency. The Journal of Clinical Endocrinology and Metabolism, JCEM. 2015; 100(7): E1005–E1013.
- Strushkevich N, Usanov SA, Plotnikov AN, et al. Structural analysis of CYP2R1 in complex with vitamin D3. Journal of Molecular Biology, JMB. 2008; 380: 95–106.
- Venselaar H, Te Beek TA, Kuipers RK, et al. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BioMed Central Bioinformatics, BMC Bioinformatics. 2010; 11: 548.
- Mutair ANAI, Nasrat GH, Russell DW. Mutation of the CYP2R1 Vitamin D 25-Hydroxylase in a Saudi Arabian Family with Severe Vitamin D Deficiency. The Journal of Clinical Endocrinology & Metabolism, JCEM. 2012; 97(10): E2022–E2025.
- Afaf AlSagheir, Faiqa Imtiaz, Sarah Bakhamis, et al. Clinical and Molecular Characterization of 25-Hydroxylase Deficiency in Saudi Patients. European Society for Paediatric Endocrinology Abstracts, ESPE Abstracts. 2016:86 P-P1-116.
- Arnaud Molin, Arnaud Wiedemann, Nick Demers, et al. Vitamin D–Dependent Rickets Type 1B (25-Hydroxylase Deficiency): A Rare Condition or a Misdiagnosed Condition?. Journal of Bone and Mineral Research, JBMR. 2017; 32: 1–7.