
Genetic dissection of Chiari malformation type 1 using endophenotypes and stratification
Aintzane Urbizu1, Tahir N. Khan2, Allison E. Ashley-Koch1*
1Duke Molecular Physiology Institute, Duke University Medical Center, Durham, NC, USA
2Duke Center for Human Disease Modeling, Duke University Medical Center, Durham, NC, USA
Abstract
Chiari malformation type 1 is a heterogeneous disease characterized by cerebellar tonsillar herniation through the foramen magnum. Symptomatology is diverse, and diagnosis and treatment are controversial. Some evidence suggests the presence of a genetic component to the disease. However, the specific genetic factors involved remain relatively unknown. Previous reviews have broadly addressed different aspects (clinical manifestations, anatomical trails, treatment) of CM-1 by itself or compared it with other types of Chiari malformation. In this mini-review, we focus our attention on the heterogeneity of this disease and its impact on the study of the genetic etiology of classic CM-1. Patient stratification strategies and endophenotypes definitions are offered to help overcome the heterogeneity.
Chiari malformation type 1
Chiari malformation type 1 (CM-1) is the most prevalent form of the “Chiari malformations”, and is characterized by a downward herniation of the caudal part of the cerebellum through the foramen magnum into the upper cervical region (Figure 1). It is a very heterogeneous disease whose current diagnosis relies on an imaging observation of cerebellar tonsil herniation (TH) of at least 3-5mm below the foramen magnum1,2. This TH is usually attributed to a reduced size of the posterior cranial fossa (PCF) (classic CM-1), although other mechanisms may be involved (see below). That is, the subsequent smaller cranial space leads to overcrowded neural structures and the herniation of the cerebellum through the foramen magnum. This results in a direct compression of the neural tissue at the craniovertebral junction and, often, cerebrospinal fluid (CSF) disturbances (decreased velocity and elevated impedance), that can cause other related conditions such as syringomyelia or secondary hydrocephalus3,4.
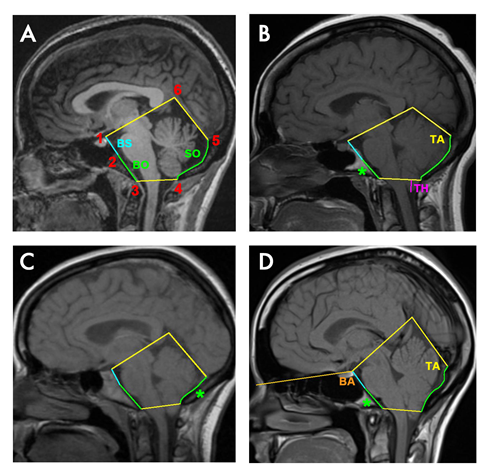
Figure 1
Sagittal T1W1 MRI showing the PCF in four different adult women. In A) Control, B) Classic CM-1 patient with a short basioccipital bone, reduced clivus slope and smaller tentorium angle, C) Classic CM-1 patient with a short supraoccipital bone and smaller tentorium angle, and D) Classic CM-1 patient with a short basioccipital bone, reduced clivus slope and bigger tentorium angle. Cerebellum tonsillar herniation (TH) is showed in magenta in B. The occipital bone is shown in green: basioccipital (BO, from 2 to 3) and supraoccipital (SO, from 4 to 5). Basisphenoid (BS, from 1 to 2) with basioccipital results in the clivus (from 1 to 3). The asterisk shows the shorter occipital part in each patient. Tentorium angle (TA) is given by the tentorium cerebelli (from 5 to 6) and supraoccipital bone. Basal angle (BA) > 143º is considered platybasia.
CM-1 is quite heterogeneous with respect to symptomatology, epidemiology and treatment. The symptomatology presented by CM-1 patients is diverse, and its severity does not correlate with the degree of TH, with some asymptomatic cases presenting with prominent TH3,5.. The onset of symptoms generally develops gradually, however, trauma, coughing/sneezing or pregnancy can also precipitate the event3,6,7. The incidence, prevalence and distribution of CM-1 is still unclear, and estimations vary depending on the criteria used to characterize the disease: TH criterion alone, TH criterion accompanied by symptomatology or a defining mechanism of the TH. For example, in the United States (US), almost 1 % of normal adults undergoing MRI scanning have at least 5mm of TH but only about 0.01–0.04 % of adults demonstrate symptoms of CM-18. Epidemiological data from other countries is generally missing. Moreover, in the US, estimates of the prevalence based on specific subtypes and/or comorbid conditions are scarce. Thus, the true prevalence of the condition, with respect to all forms of clinical heterogeneity is unknown. The treatment varies depending on the etiology of the TH. Indeed, in classic CM-1 it also depends on the severity of symptoms, the degree of TH and the presence of other conditions (i.e. syringomyelia or scoliosis). The most common treatment for these patients is surgical PCF decompression (alone or with duraplasty), although cerebellar tonsillectomy, cervical laminectomy, suboccipital cranioplasty are also applied9,10. The goal of these surgical procedures is to decompress the foramen magnum and increase the subarachnoid space in order to avoid the impaction of the cerebellar tonsils, reestablish the CSF flow and reverse the symptoms. However, there is still not a consensus about the best procedure to follow, since not all surgeries result in improvement of symptoms and often more than one surgery is required8.
The presence of multiplex families with several CM-1 cases, the co-occurrence of CM-1 in monozygotic twins and the co-inheritance with known genetic syndromes (Table 1) strongly argue for a genetic contribution to CM-1 pathogenicity11. Despite this evidence, the precise genetic variants causative of the disease remain elusive in most cases. Several genetic studies, using different approaches (mutational analysis, whole genome linkage analysis, genetic association, expression analysis) have been performed in order to attempt to identify the genetic traits underlying this disease11-16. This work has resulted in the implication of several chromosomal regions and a number of candidate genes (Table 2). However, it is important to highlight that the major findings were achieved when the CM-1 population was stratified and analyzed in more homogenous clinical groups.
Table 1. Genetic disorders which can co-occur with CM-1.
| Disorder or Syndrome | Associated clinical anomalies | Chromosome location (gene) | OMIM | Inheritance model | Reference |
|---|---|---|---|---|---|
| Achondroplasia | Skeletal | 4p16.3 (FGFR3) | #100800 | AD & sporadic | 35 |
| Acromegaly | Endocrine | 20q13.32 (GNAS) | #102200 | Unknown | 36-38 |
| Blepharophimosis | Ophthalmic | 3q23 (FOXL2) | #110100 | AD | 39 |
| Bone marrow failure syndrome 1 | Hematologic | 4q12 (SRP72), | #614675 | AD & AR | 40 |
| 9q22 (ERCC6L2) | #615715 | ||||
| Cleidocranial dysplasia | Skeletal | 6p21.1 (RUNX2) | #119600 | AD | 41 |
| Craniometaphyseal dysplasia | Skeletal | 5p15.2 (ANKH), | #123000, | AD & AR | 42 |
| 6q22.31 (GJA1) | #218400 | ||||
| Cystic fibrosis | Exocrine | 7q31.2 (CFTR) | #219700 | AR | 43-45 |
| Epilepsy | Neurologic | 8q24 | %600131 | AD | 46 |
| Fanconi anemia | Hematologic | 16q24.3 (FANCA) | #227650 | AR | 47 |
| Furhmann syndrome | Skeletal, dermatologic & endocrine | 3p25.1 (WNT7A) | #228930 | AR | 48 |
| Goldenhar syndrome | Skeletal | 14q32 | %164210 | AD | 49 |
| Growth hormone deficiency | Endocrine | 17q23.3 (GH1) | #262400 | AD, AR & X-linked | 50-54 |
| Xq22.1 (BTK) | #173100 | ||||
| #307200 | |||||
| Hajdu–Cheney syndrome | Skeletal | 1p12 (NOTCH2) | #102500 | AD | 55 |
| Hyper IgE syndrome | Immune | 17q21.2 (STAT3) | #147060 | AD | 56 |
| Hypophosphatemic rickets | Skeletal | Xp22.2-p22.1 (PHEX) | #307800 | X-linked | 57, 58 |
| Kabuki syndrome | Miscellaneous (FF, ID, skeletal, etc.) | 12q13.12 (KMT2D) | #147920 | AD | 59, 60 |
| Klippel-Feil syndrome | Skeletal | 8q22.1 (GDF6), | #118100 | AD, AR & sporadic | 3, 24, 61 |
| 17q21.31 (MEOX1), | #214300 | ||||
| 12p13.31 (GDF3) | #613702 | ||||
| Leopard syndrome | Miscellaneous (dermatologic, etc.) | 12q24.13 (PTPN11) | #151100 | AD | 62, 63 |
| Neurofibromatosis type I | Dermatologic | 17q11.2 (NF1) | #162200 | AD | 64-70 |
| Noonan syndrome | Miscellaneous (skeletal, FF, etc.) | 12q24.13 (PTPN11) | #163950 | AD | 71 |
| Osteopetrosis | Skeletal | 11q13.2 (LRP5), | #166600, | AD, AR & X-linked | 72-74 |
| 13q14.11 (TNFS11), | #611490 | ||||
| 16p13.3 (CLCN7) | #607634 | ||||
| Paget disease of bone | Skeletal | 18q22.1 (TNFRSF11A), | #602080 | AD & AR | 75, 76 |
| 5q35 (SQSTM1), | #167250 | ||||
| 5q31 (PDB4) | %606263 | ||||
| 8q24.12 (TNFRSF11B) | #239000 | ||||
| 1q21.3 (ZNF687) | #616833 | ||||
| Pseudohypoparathyroidism type 1A | Endocrine | 20q13.32 (GNAS) | #103580 | AD | 77 |
| Renal-coloboma syndrome | Ophthalmic & renal | 10q24.3-q25.1 (PAX-2) | #120330 | AD | 78 |
| Rubenstein-Taybi syndrome | Miscellaneous (ID, skeletal, FF, etc.) | 16p13.3 (CREBBP), | #180849, | AD & sporadic | 53, 79, 80 |
| 22q13.2 (EP300) | #613684 | ||||
| Scoliosis | Skeletal | 19p13.3, | %181800 | AD | 81-85 |
| 17p11.2 | %607354 | ||||
| 8q12 | %608765 | ||||
| 9q31.2-q34.2 | %612238 | ||||
| Severe combined pituitary hormone deficiency | Endocrine | 3p11.2 (POU1F1), | #613038 | AD & AR | 53 |
| 5q35.3 (PROP1), | #262600 | ||||
| 9q34.3 (LHX3), | #221750 | ||||
| 1q25.2 (LHX4) | #262700 | ||||
| Spondylo-epiphyseal dysplasia tarda | Skeletal | Unknown | #271600 | AD, AR & X-linked | 86 |
| Townes-Brocks syndrome | Skeletal & gastrointestinal | 16q12.1 (SALL1) | #107480 | AD | 87 |
| Velocardiofacial syndrome | Miscellaneous (skeletal, neurologic, etc.) | 22q11(TBX1) | #192430 | AD | 88 |
| William's syndrome | Miscellaneous (ID, FF, cardiovascular, etc.) | 7q11.2 (Deletion of aprox 28 genes including: LIMK1, RFC2, WBSCR1, WBSCR2) | #194050 | AD | 89, 90 |
AD: autosomal dominant inheritance, AR: autosomal recessive inheritance, FF: facial features, ID: intellectual disability
Table 2. List of the candidate genes suggested for CM-1 from genetic studies performed.
|
Chromosome location |
Gene | OMIM | Encodes for / Involved in | Type of study | Reference |
|---|---|---|---|---|---|
| 1p36 | RUNX3 | *600210 | Transcription factor / Segmentation body in Drosophila | Expression | 16 |
| 1q25.2 | LHX4 | *602146 | Transcription factor / Differentiation & development of pituitary gland | Linkage analysis | 20 |
| 3p21.31 | PTH1R | *168468 | Receptor parathyroid hormone & parathyroid hormone / Chondrodysplasias & enchodromatosis | Expression | 16 |
| 3p22 | TGFBR2 | *190182 | Transmembrane protein / Transcription genes related cell proliferation | Expression | 16 |
| 3p22-p21.2 | RPL14 | Ribosomal protein | eQTL | 15 | |
| 3q26.2 | RPL22L1 | Ribosomal protein | eQTL | 15 | |
| 5q14.2 | RPS23 | *603683 | Ribosomal protein | eQTL | 15 |
| 5q32 | CDX1 | *600746 | Transcription factor / Anterior-posterior regional identify | Association | 13 |
| 6p21 | RUNX2 | *600211 | Transcription factor / Osteoblastic differentiation & skeletal morphogenesis | Expression | 16 |
| 6p21.3 | NOTCH4 | *164951 | Transmembrane protein / Chondrocyte Proliferation & maduration | Expression | 16 |
| 8q12 | RPS20 | *603682 | Ribosomal protein | eQTL | 15 |
| 8q22.1 | GDF6 | *601147 | Member of BMP family and the TGF-beta superfamily/ Bone formation | Linkage analysis | 14 |
| 9q34.3 | NOTCH1 | *190198 | Transmembrane protein / Proliferation & maduration of chondrocytes | Expression | 16 |
| 11q23.3 | ETS1 | *164720 | Transcription factor / Osteoblast differentiation & bone formation | Expression | 16 |
| 12p11.21 | IPO8 | *605600 | Ras-related small GTP-binding protein / Osteoblast differentiation | eQTL | 15 |
| 12p13.31 | GDF3 | *606522 | Ligand of TGF-beta protein / Ocular & skeletal development | Linkage analysis | 14 |
| Klippel-Feil syndrome | Skeletal | 8q22.1 (GDF6), | #118100 | AD, AR & sporadic | 3, 24, 61 |
| 12q13 | RPS26 | *603701 | Ribosomal protein | eQTL | 15 |
| 12q13.11 | COL2A1 | +120140 | Collagen / Cartilage & the vitreous humor of the eye | Expression | 16 |
| 13q12 | FLT1 | *165070 | Receptor tyrosine kinases / Angiogenesis & vasculogenesis | Association | 13 |
| 14q21 | RPL36AL | *180469 | Ribosomal protein L36A-like | eQTL | 15 |
| 15q21.3 | ALDH1A2 | *603687 | Aldehyde dehydrogenase protein / Mesoderm differentiation & somitogenesis | Association | 13 |
| 15q21.1 | FBN1 | *134797 | Mature extracellular matrix glycoprotein / Connective tissue | Linkage analysis | 12 |
| 16p12.3 | XYLT1 | *608124 | Xylosyltransferase enzyme / Ossification | eQTL | 15 |
| 17q24.2 | PRKAR1A | *188830 | Protein kinase / Associated with genetic disorder of bone growth | eQTL | 15 |
| 21q22.2 | ETS2 | *164740 | Transcription factor / Osteoblast differentiation & bone formation | Expression | 16 |
| 22q13.1 | ATF4 | *604064 | Transcription factor / Osteoblast differentiation | Linkage analysis | 20 |
| 22q13.2 | EP300 | *602700 | Histone acetyltransferase / Cell proliferation & differentiation | Linkage analysis | 20 |
Strategies to consider in future genetic studies of CM-1
In a heterogeneous condition like CM-1, it is essential to properly characterize the patients, not only to establish a good diagnosis, but also to decide the best treatment to follow and to perform studies that help to improve the etiologic knowledge of the disease. Although TH is widely used in the diagnosis of CM-1, it is not heritable or completely correlated with symptomatic disease12,16. Since TH seems to be secondary in CM-1, other factors should be considered in order to successfully identify the genetic etiology of CM-1.
TH mechanism
As mentioned above, it has been generally assumed that TH was a consequence of a hypoplasic PCF due to a shortening of the occipital bone caused by an insufficiency during paraxial mesoderm development17. However, other cranial constriction mechanisms, such as premature closure of the cranial sutures (craniosynostosis), can also produce a reduced PCF with a smaller occipital bone; in addition, the reduced PCF is not the only explanation for the origin of TH. Other mechanisms such as cranial settling, occipitoatlantoaxial joint instability, spinal cord tethering, intracranial hypertension and intraspinal hypotension, can lead to TH18. For some of these mechanisms, different causal genetic pathways have been described. For example, craniosynostosis with TH (OMIM: #101200, #304110, #123500, #101600, #182212) can be produced by mutations in the FGFR1, FGFR2, EFNB1 and SKI genes; and connective tissue disorders with TH (OMIM: #601776, #615539, #609192, #610168, #613795, #615582, #154700) have been associated with mutations in the DSE, CHST14, TGFBR1, TGFBR2, SMAD3, TGFB3 and FBN1 genes.
In classic CM-1, most candidate genes have been related to the different stages of occipital bone development, including the paraxial mesoderm development, somite and sclerotome formation, chondrogenesis and osteogenesis (Table 2)13,15,16,19,20.
Stratifying according to the TH mechanism (presence or absence of history of connective tissue disorders), Markunas et al. observed that the evidence for genetic linkage in several chromosomal regions increased significantly and led to the identification of two missense mutations with incomplete penetrance in the GDF6 gene in two independent classic CM-1 families14 (Table 2).
PCF MRI-morphometric traits as endophenotypes
Although TH herniation is not heritable12,16, other PCF traits are, and the use of these heritable traits are more likely to aid in the identification of the underlying genes.
PCF morphometric characteristics are different in CM-1 patients according to the mechanism of TH18. In addition, although a hyploplastic PCF is a common trait for classic CM-1 patients, the regions of the PCF are not equally affected16,13,20. Usually the PCF is shallower, as a result of the shorter occipital bone, and often the slope of the clivus is reduced, resulting in a predisposition to platybasia. Most studies demonstrated the basilar part of the occipital bone (basioccipital) or clivus (when the sphenoccipital synchondrosis is not visible) is significantly reduced6,21,22. However, for some cohorts this reduction seems to be more significant in the supraoccipital part23,24, or present for both regions4. There are also conflicting reports about the magnitude of the tentorium angle in patients compared to control cohorts3,22 (Figure 1).
Since the formation and development of the occipital bone is intricate, a morphometric analysis of the PCF based on MRI is essential in classic CM-1 patients. Depending on which part of the occipital bone is abnormal (shorter and/or present with different slope), the genes involved may be different. The occipital bone is formed from the fusion of the first four somites. However, the parts of these somites that resegment to form the sclerotomes are different for each part of the bone25. The basilar part fuses with the basisphenoid bone at the sphenoocipital synchondrosis (which derives from neural crests cells and the mesoderm, and its closure finishes at the age of 20 years)8; but the boundaries of the supraoccipital part are given by the interparietal-supraoccipital, lamboid and occipito-mastoid sutures8,26. In addition, the occipital bone has both membranous and cartilaginous types of ossification, and the number of ossification centers for each part is different8,26.
After considering the PCF traits presented in CM-1 patients, Urbizu et al. identified four genetic variants (located in the genes ALDH1A2, CDX1 and FLT1) to be associated with adult classic CM-1, and two of them also associated with the slope of the clivus31(Table 2). Using MRI endophenotypes defining two different “shapes” of PCF, Markunas et al. identified different levels of expression in genes related with dorso-ventral axis formation (ETS1, ETS2, NOTCH4), ribosome, spliceosome and proteasome in pediatric classic CM-1 patients16 (Table 2). These initial findings support the complex genetic basis of the PCF development, and the genetic heterogeneity of CM-1.
Co-occurrence with other genetic disorders
Another approach to identifying genes for CM-1 is to stratify CM-1 according to co-occurring genetic disorders, either by syndrome or according to the associated clinical anomalies (skeletal, hematologic, ophthalmic, endocrine, exocrine, dermatologic, neurologic or immune) (Table 1). Importantly, some of these different disorders may be related since: 1) they are caused by the same gene/locus (i.e. mutations in GNAS gene have been identified in patients with acromegaly and Pseudohypoparathyroidism type 1A), 2) some of these genes interact (i.e. CREBBP and EP300 are genes that cause Rubenstein-Taybi syndrome, and are involved in chondrocyte differentiation; they interact with LHX4 which is associated with severe combined pituitary hormone deficiency, also described in CM-1 patients), and 3) some of these genes are in the same pathway (i.e. Noonan syndrome and Neurofibromatosis type 1 are caused by alterations in Ras pathway genes).
Familial or sporadic forms of CM-1
The majority of CM-1 cases are believed to be sporadic. However, familial cases (presenting autosomal recessive and autosomal dominant inheritance with incomplete penetrance) have also been reported12. Thus, it is possible that the presence or absence of family history is associated with different genes and disease pathogenesis. For example, in some diseases such as Fronto Temporal Dementia or Amyothropic Lateral Sclerosis, different genes, and even different pathways, are affected depending if the cases are familial or sporadic27. The fact that some CM-1 families present different inheritance patterns also suggests that there is more than one gene involved in the disease.
Age of symptom onset
Traditionally, CM-1 has been considered an adult form of the Chiari malformations since the onset of the symptoms usually occurs during the second or third decade of life. More recently, because of the increased use of MRI diagnosis and clinical awareness, this perception is changing, with an increasing number of pediatric cases being reported10,28,29 . Both pediatric and adult CM-1 patients have TH in common. However, the symptomatology, comorbidities and gender proportion are not exactly the same. For example, and although the precise estimate is still unknown, in adults a higher incidence is generally observed for females, while in pediatric cases this incidence is more evenly distributed among sexes3,10,13,18, 19,30.These differences could indicate that pediatric and adult onset cases are different forms of the disease. There are many examples of other diseases where age of onset is important. In Alzheimer disease, different genetic variants are involved in the early (younger than 65 years old) and late onset forms of the disease31.
Gender
The fact that in CM-1 adult form is more common in females, suggests the possibility of sexual-dimorphism. Gender effects can manifest in the presentation of the disease, associated symptoms, prevalence, or age of onset. These differences are also seen in CM-1 as we have described above. Sexual-dimorphism has been observed in other complex human diseases such as cardiovascular disease, asthma, autoimmune diseases, some neurological and psychiatric disorders, as well as some common birth defects and cancers32. Many complex human diseases exhibit sex and age differences in gene expression where common variants may alter gene expression and influence disease susceptibility or its progression33. For CM-1, the alteration in the occipital bone could be produced at different developmental time points, from embryonic stages until the completion of bone development. Importantly, hormones (parathyroid hormone, growth hormone), including sexual hormones (estrogen, testosterone), are involved in bone growth34. Interestingly, some disorders that co-occur with CM-1 are caused by endocrine alterations (Table 1).
Conclusion
In conclusion, there remains little known about the genetic component involved in the etiology of CM-1. There is a tremendous need to replicate the published studies and validate the findings in independent cohorts, as well as to consider the effect of these stratification strategies on proposed candidate genes. We also cannot discard other genomic mechanisms, such as regulatory variation and epigenetic modifications, as additional contributors to disease etiology.
Thus, other genomic approaches may be needed, such as epigenetic analyses and next generation sequencing, which have not yet been applied to CM-1. Ultimately, however, a better understanding of meaningful clinical stratifications will be required to identify more genetically homogeneous subsets in order to find the causative genetic variants.
Acknowledgements
A.U. is the recipient of a Postdoctoral Fellowship from Fundación Ramón Areces (Spain). We would like to thank Conquer Chiari for providing the images in Figure 1.
References
- Aboulezz AO, Sartor K, Geyer CA, et al. Position of cerebellar tonsils in the normal population and in patients with Chiari malformation: a quantitative approach with MR imaging. J Comput Assist Tomogr. 1985; 9: 1033-1036.
- Barkovich AJ, Wippold FJ, Sherman JL, et al. Significance of cerebellar tonsillar position on MR. AJNR Am J Neuroradiol. 1986; 7: 795-799.
- Milhorat TH, Chou MW, Trinidad EM, et al. Chiari I malformation redefined clinical and radiographic findings for 364 symptomatic patients. Neurosurgery. 1999; 44: 1005-1017.
- Shaffer N, Martin BA, Rocque B, et al. Cerebrospinal fluid flow impedance is elevated in Type I Chiari malformation. J Biomech Eng. 2014; 136: 021012.
- Meadows J, Kraut M, Guarnieri M, et al. Asymptomatic Chiari Type I malformations identified on magnetic resonance imaging. J Neurosurg. 2000; 92: 920-926.
- Aydin S, Hanimoglu H, Tanriverdi T, et al. Chiari type I malformations in adults: a morphometric analysis of the posterior cranial fossa. Surg Neurol. 2005; 64: 237-241. discussion 241.
- Wan MJ, Nomura H, Tator CH. Conversion to symptomatic Chiari I malformation after minor head or neck trauma. Neurosurgery. 2008; 63: 748-753. discussion 753.
- Tubbs RS, Oakes WJ. The Chiari malformations. Springer New York United States. 2013.
- Zhang ZQ, Chen YQ, Chen YA, et al. Chiari I malformation associated with syringomyelia a retrospective study of 316 surgically treated patients. Spinal Cord. 2008; 46: 358-363.
- Tubbs RS, Beckman J, Naftel RP, et al. Institutional experience with 500 cases of surgically treated pediatric Chiari malformation Type I. Journal of neurosurgery Pediatrics. 2011; 7: 248-256.
- Speer MC, Enterline DS, Mehltrette L, et al. Chiari Type I Malformation With or Without Syringomyelia Prevalence and Genetics. Journal of Genetic Counseling. 2003; 12: 297-311.
- Boyles AL, Enterline DS, Hammock PH, et al. Phenotypic definition of Chiari type I malformation coupled with high-density SNP genome screen shows significant evidence for linkage to regions on chromosomes 9 and 15. Am J Med Genet A. 2006; 140: 2776-2785.
- Urbizu A, Toma C, Poca MA, et al. Chiari Malformation Type I A Case-Control Association Study of 58 Developmental Genes. PLos ONE. 2013; 8: e57241.
- Markunas CA, Soldano K, Dunlap K, et al. Stratified whole genome linkage analysis of Chiari type I malformation implicates known Klippel-Feil syndrome genes as putative disease candidates. PloS one. 2013; 8: e61521.
- Lock EF, Soldano KL, Garrett ME, et al. Joint eQTL assessment of whole blood and dura mater tissue from individuals with Chiari type I malformation. BMC Genomics. 2015; 16: 11.
- Markunas CA, Lock E, Soldano K, et al. Identification of Chiari Type I Malformation subtypes using whole genome expression profiles and cranial base morphometrics. BMC medical genomics. 2014; 7: 39.
- Marin Padilla M, Marin Padilla TM. Morphogenesis of experimentally induced Arnold Chiari malformation. J Neurol Sci. 1981; 50: 29-55.
- Milhorat TH, Nishikawa M, Kula RW, et al. Mechanisms of cerebellar tonsil herniation in patients with Chiari malformations as guide to clinical management. Acta Neurochir (Wien). 2010; 152: 1117-1127.
- Speer MC, George TM, Enterline DS, et al. A genetic hypothesis for Chiari I malformation with or without syringomyelia. Neurosurg Focus. 2000; 8: E12.
- Markunas CA, Enterline DS, Dunlap K, et al. Genetic evaluation and application of posterior cranial fossa traits as endophenotypes for Chiari type I malformation. Annals of human genetics. 2014; 78: 1-12.
- Noudel R, Jovenin N, Eap C, et al. Incidence of basioccipital hypoplasia in Chiari malformation type I comparative morphometric study of the posterior cranial fossa. Clinical article. J Neurosurg. 2009; 111: 1046-1052.
- Urbizu A, Poca MA, Vidal X, et al. MRI based morphometric analysis of posterior cranial fossa in the diagnosis of chiari malformation type I. Journal of neuroimaging official journal of the American Society of Neuroimaging. 2014; 24: 250-256.
- Dagtekin A, Avci E, Kara E, et al. Posterior cranial fossa morphometry in symptomatic adult Chiari I malformation patients Comparative clinical and anatomical study. Clin Neurol Neurosurg. 2011; 113: 399-403.
- Nishikawa M, Sakamoto H, Hakuba A, et al. Pathogenesis of Chiari malformation: a morphometric study of the posterior cranial fossa. J Neurosurg. 1997; 86: 40-47.
- Pang D, Thompson DN. Embryology and bony malformations of the craniovertebral junction. Childs Nerv Syst. 2011; 27: 523-564.
- Bernard S, Loukas M, Rizk E, et al. The human occipital bone review and update on its embryology and molecular development. Childs Nerv Syst. 2015; 31: 2217-2223.
- Al-Chalabi A, Jones A, Troakes C, et al. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012; 124: 339-352.
- Greenlee JD, Donovan KA, Hasan DM, et al. Chiari I malformation in the very young child the spectrum of presentations and experience in 31 children under age 6 years. Pediatrics. 2002; 110: 1212-1219.
- Aitken LA, Lindan CE, Sidney S, et al. Chiari type I malformation in a pediatric population. Pediatric neurology. 2009; 40: 449-454.
- Strahle J, Muraszko KM, Kapurch J, et al. Chiari malformation Type I and syrinx in children undergoing magnetic resonance imaging. Journal of neurosurgery Pediatrics. 2011; 8: 205-213.
- Chouraki V, Seshadri S. Genetics of Alzheimer's disease. Adv Genet. 2014; 87: 245-294.
- Morrow EH. The evolution of sex differences in disease. Biol Sex Differ. 2015; 6: 5.
- Yao C, Joehanes R, Johnson AD, et al. Sex and age interacting eQTLs in human complex diseases. Hum Mol Genet. 2014; 23: 1947-1956.
- Karsenty G. The genetic transformation of bone biology. Genes Dev. 1999; 13: 3037-3051.
- Nakai T, Asato R, Miki Y, et al. A case of achondroplasia with downward displacement of the brain stem. Neuroradiology. 1995; 37: 293-294.
- Lemar HJ, Perloff JJ, Merenich JA. Symptomatic Chiari-I malformation in a patient with acromegaly. South Med J. 1994; 87: 284-285.
- Hara M, Ichikawa K, Minemura K, et al. Acromegaly associated with Chiari-I malformation and polycystic ovary syndrome. Intern Med. 1996; 35: 803-807.
- Manara R, Bommarito G, Rizzati S, et al. Herniation of cerebellar tonsils in acromegaly prevalence pathogenesis and clinical impact. Pituitary. 2013; 16: 122-130.
- Paquis P, Lonjon M, Brunet M, et al. Chiari Type I malformation and syringomyelia in unrelated patients with blepharophimosis. Report of two cases. J Neurosurg. 1998; 89: 835-838.
- Akiyama K, Koh K, Mori M, et al. Association between Chiari malformation and bone marrow failure myelodysplastic syndrome. Br J Haematol. 2013; 163: 411-412.
- Vari R, Puca A, Meglio M. Cleidocranial dysplasia and syringomyelia. Case report J Neurosurg Sci. 1996; 40: 125-128.
- Cai C, Zhang Q, Shen C, et al. Chiari malformation caused by craniometaphyseal dysplasia: case report and review of literature. European journal of pediatric surgery official journal of Austrian Association of Pediatric Surgery . Zeitschrift fur Kinderchirurgie. 2008; 18: 198-201.
- Needleman JP, Panitch HB, Bierbrauer KS, et al. Chiari type I malformation in children and adolescents with cystic fibrosis. Pediatric pulmonology. 2000; 30: 490-492.
- Rakheja D, Xu Y, Burns DK, et al. Cystic fibrosis and Chiari type I malformation: autopsy study of two infants with a rare association. Pediatric and developmental pathology the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society. 2003; 6: 88-93.
- Patel AJ, Raol VH, Jea A. Rare association between cystic fibrosis Chiari I malformation and hydrocephalus in a baby a case report and review of the literature. Journal of medical case reports. 2011; 5: 366.
- Granata T, Valentini LG. Epilepsy in type 1 Chiari malformation. Neurol Sci. 2011; 32 Suppl 3: S303-306.
- Amalnath D, Subramanian R, Swaminathan R, et al. Incidental detection of Chiari malformation in Fanconi anaemia. Br J Haematol. 2012; 154.
- Pfeiffer RA, Stoss H, Voight HJ, et al. Absence of fibula and ulna with oligodactyly contractures right angle bowing of femora abnormal facial morphology cleft lip palate and brain malformation in two sibs a possibly new lethal syndrome. Am J Med Genet. 1988; 29: 901-908.
- Mesiwala AH, Shaffrey CI, Gruss JS, et al. Atypical hemifacial microsomia associated with Chiari I malformation and syrinx further evidence indicating that chiari I malformation is a disorder of the paraaxial mesoderm Case report and review of the literature. J Neurosurg. 2001; 95: 1034-1039.
- Hamilton J, Blaser S, Daneman D. MR imaging in idiopathic growth hormone deficiency. AJNR Am J Neuroradiol. 1998; 19: 1609-1615.
- Murphy RL, Tubbs RS, Grabb PA, et al. Chiari I malformation and idiopathic growth hormone deficiency in siblings: report of three cases. Childs Nerv Syst. 2007; 23: 1221-1223.
- O'Grady MJ, Cody D. Symptomatic Chiari 1 malformation after initiation of growth hormone therapy. J Pediatr. 2011; 158: 686.
- Marzuillo P, Grandone A, Coppola R, et al. Novel cAMP binding protein-BP (CREBBP) mutation in a girl with Rubinstein-Taybi syndrome GH deficiency Arnold Chiari malformation and pituitary hypoplasia. BMC Med Genet. 2013; 14: 28.
- Lindsay R, Feldkamp M, Harris D, et al. Utah Growth Study growth standards and the prevalence of growth hormone deficiency. J Pediatr. 1994; 125: 29-35.
- Sawin PD, Menezes AH. Basilar invagination in osteogenesis imperfecta and related osteochondrodysplasias medical and surgical management. J Neurosurg. 1997; 86: 950-960.
- Freeman AF, Holland SM. The hyper-IgE syndromes. Immunol Allergy Clin North Am. 2008; 28: 277-291. viii.
- Caldemeyer KS, Boaz JC, Wappner RS, et al. Chiari I malformation association with hypophosphatemic rickets and MR imaging appearance. Radiology. 1995; 195: 733-738.
- Kuether TA, Piatt JH. Chiari malformation associated with vitamin D resistant rickets case report. Neurosurgery. 1998; 42: 1168-1171.
- McGaughran J, Aftimos S, Jefferies C, et al. Clinical phenotypes of nine cases of Kabuki syndrome from New Zealand. Clin Dysmorphol. 2001; 10: 257-262.
- Ciprero KL, Clayton-Smith J, Donnai D, et al. Symptomatic Chiari I malformation in Kabuki syndrome. Am J Med Genet A. 2005; 132A: 273-275.
- Ritterbusch JF, McGinty LD, Spar J, et al. Magnetic resonance imaging for stenosis and subluxation in Klippel Feil syndrome. Spine (Phila Pa 1976). 1991; 16: S539-541.
- Agha A, Hashimoto K. Multiple lentigines Leopard syndrome with Chiara I malformation. J Dermatol. 1995; 22: 520-523.
- Beier AD, Barrett RJ, Burke K, et al. Leopard syndrome and Chiari type I malformation a case report and review of the literature. Neurologist. 2009; 15:37-39.
- Afifi AK, Dolan KD, Van Gilder JC, et al. Ventriculomegaly in neurofibromatosis-1 Association with Chiari type I malformation. Neurofibromatosis. 1988; 1: 299-305.
- Dooley J, Vaughan D, Riding M, et al. The association of Chiari type I malformation and neurofibromatosis type 1. Clin Pediatr (Phila). 1993; 32: 189-190.
- Parkinson D, Hay R. Neurofibromatosis. Surgical neurology. 1986; 25: 109-113.
- Hara H, Arakawa H. Coexistence of neurofibromatosis 1 and chiari type I malformation: an unusual association. The Journal of dermatology. 2005; 32: 34-37.
- Tubbs RS, Rutledge SL, Kosentka A, et al. Chiari I malformation and neurofibromatosis type 1. Pediatr Neurol. 2004; 30: 278-280.
- Felicio AC, Godeiro Junior Cde O, Borges V, et al. Hemifacial spasm in a patient with neurofibromatosis and Arnold-Chiari malformation: a unique case association. Arq Neuropsiquiatr. 2007; 65:855-857.
- Santos Garcia D, Cabanillas M, Suarez Dono I, et al. Type 1 neurofibromatosis and Arnold-Chiari malformation. Actas dermo sifiliograficas. 2009; 100: 820-822.
- Ejarque I, Millan Salvador JM, Oltra S, et al. Arnold-Chiari malformation in Noonan syndrome and other syndromes of the RAS/MAPK pathway. Rev Neurol. 2015; 60: 408-412.
- Jamjoom AA, Jamjoom BA, Waliuddin AR, et al. Lessons from a case of osteopetrosis oxycephaly and Chiari type I malformation a case report. Cases journal. 2009; 2: 6787.
- Kulkarni ML, Marakkanavar SN, Sushanth S, et al. Osteopetrosis with Arnold Chiari malformation type I and brain stem compression. Indian journal of pediatrics. 2007; 74: 412-415.
- Dlouhy BJ, Menezes AH. Osteopetrosis with Chiari I malformation presentation and surgical management. Journal of neurosurgery Pediatrics. 2011; 7: 369-374.
- Iglesias Osma C, Gomez Sanchez JC, Suquia Mugica B, et al. Paget's disease of bone and basilar impression associated with an Arnold-Chiari type-1 malformation. Anales de medicina interna. 1997; 14: 519-522.
- Richards PS, Bargiota A, Corrall RJ. Paget's disease causing an Arnold-Chiari Type 1 malformation: radiographic findings. AJR Am J Roentgenol. 2001; 176: 816-817.
- Martinez Lage JF, Guillen Navarro E, Lopez Guerrero AL, et al. Chiari type 1 anomaly in pseudohypoparathyroidism type Ia: pathogenetic hypothesis. Child's nervous system ChNS official journal of the International Society for Pediatric. Neurosurgery. 2011; 27: 2035-2039.
- Schimmenti LA, Shim HH, Wirtschafter JD, et al. Homonucleotide expansion and contraction mutations of PAX2 and inclusion of Chiari 1 malformation as part of renal coloboma syndrome. Hum Mutat. 1999; 14: 369-376.
- Wojcik C, Volz K, Ranola M, et al. Rubinstein Taybi syndrome associated with Chiari type I malformation caused by a large 16p13.3 microdeletion: a contiguous gene syndrome. Am J Med Genet A. 2010; 152A: 479-483.
- Parsley L, Bellus G, Handler M, et al. Identical twin sisters with Rubinstein Taybi syndrome associated with Chiari malformations and syrinx. Am J Med Genet A. 2011; 155A: 2766-2770.
- Nagib MG. An approach to symptomatic children ages 4-14 years with Chiari type I malformation. Pediatric neurosurgery. 1994; 21: 31-35.
- Park JK, Gleason PL, Madsen JR, et al. Presentation and management of Chiari I malformation in children. Pediatric neurosurgery. 1997; 26: 190-196.
- Eule JM, Erickson MA, O'Brien MF, et al. Chiari I malformation associated with syringomyelia and scoliosis: a twenty-year review of surgical and nonsurgical treatment in a pediatric population. Spine (Phila Pa 1976). 2002; 27: 1451-1455.
- Tubbs RS, Doyle S, Conklin M, et al. Scoliosis in a child with Chiari I malformation and the absence of syringomyelia case report and a review of the literature. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2006; 22: 1351-1354.
- Brockmeyer DL. Editorial Chiari malformation Type I and scoliosis: the complexity of curves. J Neurosurg Pediatr. 2011; 7: 22-23; discussion 23-24.
- Gripp KW, Nicholson L, Magram G, et al. Chiari malformation and tonsillar ectopia in twin brothers and father with autosomal dominant spondylo-epiphyseal dysplasia tarda. Skeletal Radiol. 1997; 26: 131-133.
- Kohlhase J. Townes Brocks Syndrome In GeneReviews(R) (Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, eds.). Seattle (WA), 1993.
- Hultman CS, Riski JE, Cohen SR, et al. Chiari malformation, cervical spine anomalies and neurologic deficits in velocardiofacial syndrome. Plastic and reconstructive surgery. 2000; 106: 16-24.
- Mercuri E, Atkinson J, Braddick O, et al. Chiari I malformation in asymptomatic young children with Williams syndrome clinical and MRI study. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society. 1997; 1: 177-181.
- Pober BR, Filiano JJ. Association of Chiari I malformation and Williams syndrome. Pediatric neurology. 1995; 12: 84-88.