
Myelinosome-driven secretion: Non-catabolic management of misfolded proteins - Lessons from the Sertoli cells
Marina G. Yefimova1,2 and Nicolas Bourmeyster1*
1Université de Poitiers/CNRS, Laboratoire Signalisation et Transports Ioniques Membranaires, 1 rue Georges Bonnet, F-86022 Poitiers, France
2Sechenov Institute of Evolutionary Physiology and Biochemistry, Russian Academy of Sciences, 44 Maurice Thorez Prospekt, 194233 Saint Petersburg, Russia
Abstract
Myelinosomes are pleomorphic structures with huge diversity of form, specific hallmark of “lysosomal storage diseases”, but also present in other inborn errors of metabolism such as aggregation diseases HD and CF. In the original work “Myelinosomes act as natural secretory organelles to prevent accumulation of aggregate-prone mutant Huntingtin and CFTR” Yefimova et al., describe an unexpected role of myelinosomes as key regulators of proteostasis in the somatic Sertoli cells from the testis. By releasing myelinosomes loaded with undegraded misfolded proteins, Sertoli cells demonstrate a new aspect of protein quality control mechanism unrelated to catabolic degradation.
Definition
The term “myelinosomes” was introduced by electron microscopy investigators to describe the markedly electron-dense osmiophile membranes, showing a stacked, reticulated or whorled arrangement1. While extremely rare in a variety of normal cells, intracellular myelinosomes arise in pathological situations caused by genetic or environmental factors1. Various names have been given to these organelles, which are also known as multimembranous bodies, multilamellar bodies, myelin bodies, myelin figures, myelin-like organelles, membranous cytoplasmic bodies, and zebra bodies. Generally, these particles exhibit acid phosphatase activity, classically attributed to lysosome organelles1.
Myelinosomes are detected in aggregation diseases
These electron-dense osmiophile membranes are found in greatest number in primary and secondary lysosomal storage disorders (LSD), the former caused by mutations in genes encoding lysosomal enzyme proteins and related co-factors2, the later resulting from the impairment of fusion of transient storage endocytic or autophagic organelle with lysosome3. In LSD myelinosomes are considered as “storage” organelles along with autolysosomes, endolysosomes and late endosomes. Interestingly, myelinosomes from secondary LDS (such as Niemann-Pick type C1 syndrome) are more basic (pH 5,5 - 6,0) and are devoid of ubiquitous lysosomal protease cathepsin D activity3.
While in lesser extent comparing to LSD, intracellular myelinosomes are present in aggregation disorders Huntington disease (HD) and cystic fibrosis (CF)4-6, which particularly target CNS and lung respectively. HD is caused by the expansion of CAG triplet in the gene coding for ubiquitous protein Huntingtin (Htt). The mutation results in generation of long polyglutamine track in popypeptide chain of Htt, rendering it prone to form insoluble aggregates7. CF is caused by the deletion of three nucleotides coding for phenylalanine at the position 508 of cystic fibrosis transmembrane regulator (CFTR) protein, an ubiquitously expressed ion channel, involved in the transport of chloride ions across cell membranes8. This mutation also predisposes mutant F508delCFTR protein to aggregation9. Being resistant to degradation by cellular lysosome-dependent (Autophagy-Driven-Lysosomal-Proteolysis, ADLP) and/or independent (UPS ubiquitin-proteasome-system) proteolysis machineries10,11, mHtt- or F508delCFTR-containing aggregates accumulate in targeted cells, disturb cell proteostasis and interfere with various aspects of normal cellular metabolism12.
Generally, intracellular myelinosomes are detected in aggregate-containing cells from highly affected organs (HD brain; CF lungs)5,6. Their functional significance for both HD and CF remains unclear.
Some “privileged” cells escape aggregation naturally
While in systemic level HD present a generalized pattern of aggregation, some cells of the body escape aggregation naturally. In HD these “privileged” cells are skin keratinocytes, mucosal cells and both germ and somatic Sertoli cells from the testis13. In CF bronchoalveolar epithelium, “privileged” are secretory cells responsible for surfactant production, while ciliated cells do not6. Thus, HD and CF “privileged” cells either display secretory activity to form a specific micro-environment in different tissues (Sertoli cells from seminiferous tubules, lung alveolar type II cells, skin keratinocytes, mucosal cells) or undergo strong reformation of structure with the loss of cytoplasm during differentiation process (germ cells undergoing metamorphosis from round spermatid to streamlined spermatozoa).
In this line a central question becomes: what is the mechanism supporting such “privilege”? In our work14, using secretory Sertoli cells, we demonstrate that “privilege” phenomenon relies upon myelinosome secretion, which ensures discarding potentially toxic aggregate-prone proteins out of the cells.
Myelinosomes are secretory organelles
The ability of cells to release myelinosomes has never been subjected to investigation. In our work we show for the first time that myelinosomes are secretory organelles, able to leave the cells in their entirety. It strikingly makes myelinosomes different from other intracellular secretory organelles of lysosome-related-organelles (LRO) family such as secretory granules from blood immune cells, neurons or neuroendocrine cells; lamellar bodies (LB) from lung or skin keratinocytes; melanosomes from retinal pigment epithelium; secretory lysosomes15-18. All of them discharge their content into extracellular space after the fusion with plasma membrane, but not by liberating them as intact organelles out of cell. Indeed, extracellular myelinosomes bearing mHtt were particularly frequent in R6/1 HD transgenic mice testis, in proximity of intact Sertoli and germ cells14. To our knowledge, only LRO-type secretory granules from eosinophils are known to be released as organelles during specific phagocytosis-related process termed NET (neutrophil extracellular trap)19.
The process of myelinosome maturation resembles those of lung lamellar bodies (LBs), which mature and traffic inside multivesicular bodies (MVBs). Nevertheless, MVB fusion with plasma membrane results in the discharge of LB-stored pulmonary surfactant20, while in the Sertoli cells mature myelinosomes are extruded in their entirety in extracellular spaces. This process looks alike MVB-mediated exocytosis, leading to the release of exosomes21, and is different from membrane vesicle shedding. Besides, in Sertoli cells myelinosome precursors seem to be related to ER membranes, further undergoing their maturation in MVBs.
Loading of undegraded misfolded proteins on myelinosome membranes does not result from the failure of degradation activity in Sertoli cells
The failure of lysosome degradation activity in LSD results in the accumulation of undegraded products bound to osmiophile membranes in the cytoplasm of cells from targeted organs2. As in LSD disorders, osmiophile myelinosome membranes from Sertoli cells also accumulate undegraded proteins. Nevertheless, only misfolded mHtt, F508delCFTR, or immature forms of CFTR (but not their normal counterparts) are associated with myelinosomes. This suggests an involvement of specific sorting mechanism which recognizes and addresses misfolded proteins to myelinosomes. Current knowledge of such mechanism is in its infancy. We can only state actually that loading of undegraded misfolded proteins on myelinosome membranes does not result from the failure of degradation activity in Sertoli cells, which ensures their phagocytosis and autophagy functions22-24. Indeed, phagocytosis and autophagy flux tests23,24, demonstrated that mHtt expression did not affect degradative activity of Sertoli cells. Nevertheless, we have never been able to detect any association of myelinosome-loaded mHtt and F508delCFTR with lysosomal markers as Lysotracker Red, Lamp1, Lamp2, Lamp2a, or CD63 (Yefimova et al., MS in preparation). Thus, despite of functional lysosomal machinery, Sertoli cells would rather use an alternative non-catabolic strategy to cope with aggregate-prone mHtt and F508delCFTR proteins.
In the context of LSD, the ability of Sertoli cells to bypass defective lysosomal degradative machinery as well as the use of non-catabolic pathways to maintain cell homeostasis have already been a subject of discussions25-27. In this line, of particular interest is the classical study of Chemes28, who demonstrated in Sertoli cell cytoplasm a particular subset of lysosomes, which did not exhibit acid phosphatase activity. Some of them displayed multilamellar structure28 and closely resembled myelinosomes. Therefore, it seems likely that these “neutral” multilamellar lysosomes are good candidates to serve as “garbage bags” for accumulating and further evacuating undegraded misfolded proteins out of cells.
Myelinosome-driven secretion: non-catabolic management of misfolded proteins. New aspect of protein quality control: open questions
Myelinosome-driven secretion (MDS) is a new non-catabolic PQC strategy, which coexists together with classical degradative mechanisms such as UPS, ADLP, Chaperon-mediated autophagy (CMA), MHCII in Sertoli cells (Figure 1). Sertoli cells, like probably other privileged cells, take advantage of MDS to maintain their proteostasis and vitality, avoiding accumulation of potentially toxic aggregate-prone proteins and undegraded metabolites. In fact, we detected MDS in various cell lines, as human retinal glial Müller cells, human cystic fibrosis brocho-epithelial cells or human neuroblastoma cells.
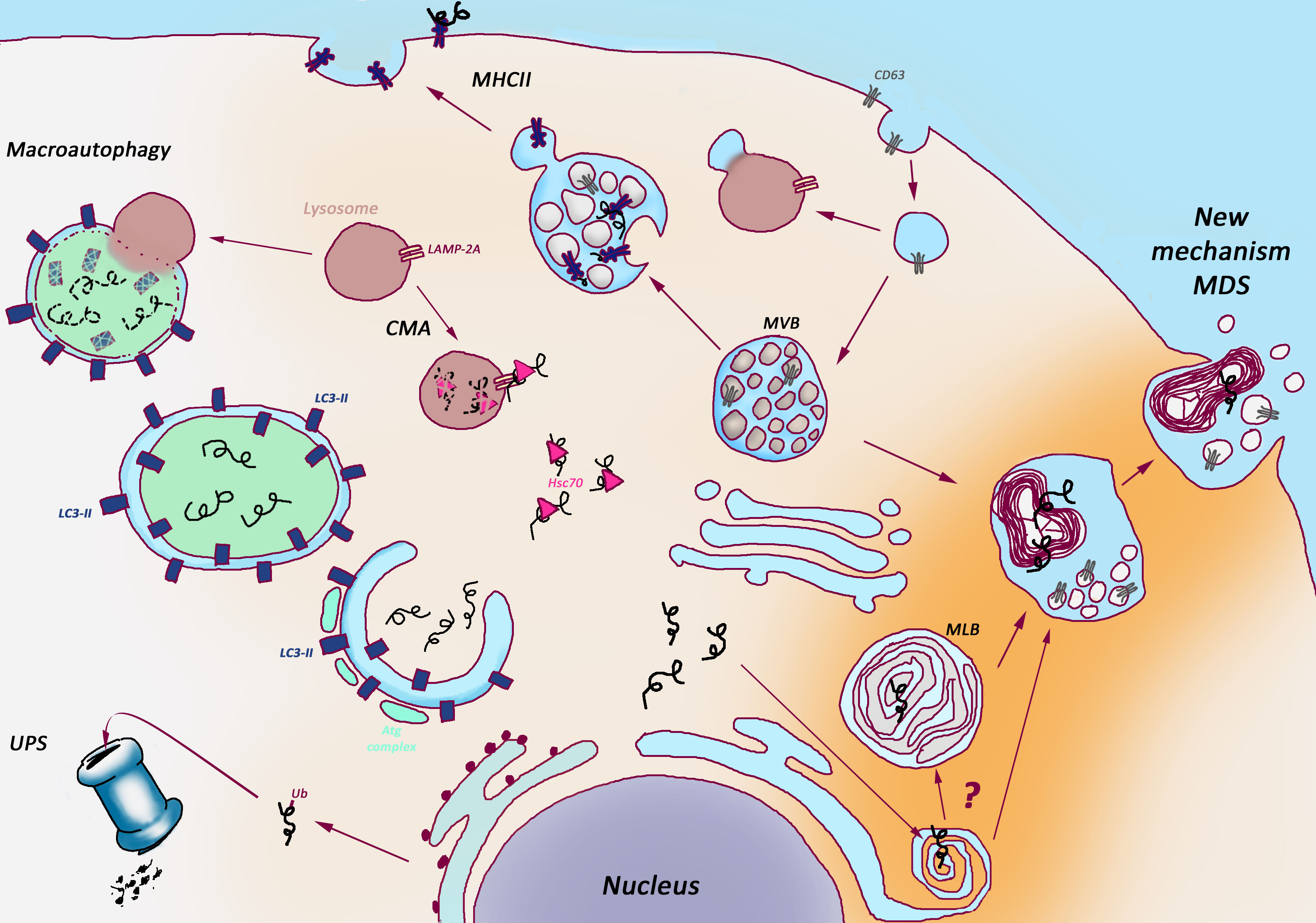
Figure 1: Myelinosome-Driven Secretion: a unique non-catabolic proteostasis pathway versus classical catabolic proteostasis pathways.
Several catabolic pathways maintain proteostasis in the cells. Ubiquitin-Proteasome System (UPS) provides the degradation of misfolded or damaged proteins tagged for elimination with a small ubiquitin protein (Ub); Autophagy delivers cytoplasmic cargo to the lysosome inside double membrane-bound vesicle (autophagosome) through both selective and non-selective mechanisms; Chaperone-Mediated-Autophagy (CMA) address the proteins with KFERQ-like sequences for degradation after translocation across the lysosomal membrane in a complex with chaperones; Major Histocompatibility Complex type II (MHCII) supports lysosome-dependent degradation and presentation on cell surface of both extracellular (phagocytosed) and intrinsic proteins in antigen-presenting cells. Non-catabolic Myelinosome-Driven Secretion (MDS) overcomes degradation stage, directly addressing misfolded proteins out of cell.
CMA: Chaperon-mediated Autophagy, MHCII: Major Histocompatibility Complex type II, MLB: MultiLamellar Bodies, MVB: MultiVesicular Bodies, UPS: Ubiquitin-Proteasome System.
Nevertheless, in the context of MDS functioning in multicellular organisms the important issues to solve are:
• What is a fate of myelinosomes extruded in extracellular spaces? Are they further addressed into biological fluids as seminal fluid, alveolar fluid, cerebrospinal fluid, lymph, and others? If so, do myelinosomes affect special functions, for example male fertility?
• Being protective mechanism for “privileged” cells, what is the impact of MDS on neighboring cells? Are myelinosomes absorbed by neighboring cells? If so, does the absorption of myelinosomes loaded with potentially toxic aggregate-prone proteins deleterious for acceptor cells? Does myelinosome transfer from cell-to-cell contribute to disease propagation as currently discussed for many neurodegenerative disorders?
• Can MDS stimulation further serve as new tool for curing LSD or aggregation diseases?
• What are fundamental physiological mechanisms supporting the equilibrium between protective versus deleterious effects of MDS?
We are only in the beginning of knowledge about MDS, and upcoming studies would bring the information concerning its role in health and disease.
References
- Ghadially FN. Ultrastructural Pathology of the Cell and Matrix Set 4th edition Boston Butterworth–Heinemann. 1997.
- Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med. 2015; 66: 471-486.
- Platt FM, Boland B, van der Spoel AC. The cell biology of disease lysosomal storage disorders the cellular impact of lysosomal dysfunction. J Cell Biol. 2012; 199: 723-734.
- Aguzzi A, O’Connor T. Protein aggregation diseases pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010; 9: 237–248.
- Chen J, Marks E, Lai B, et al. Iron accumulates in Huntington's disease neurons: protection by deferoxamine. PLoS One. 2013; 8(10):e77023. Erratum in: PLoS One. 2013; 8(11).
- Du K, Karp PH, Ackerley C, et al. Aggregates of mutant CFTR fragments in airway epithelial cells of CF lungs: new pathologic observations. J Cyst Fibros. 2015; 14: 182–193.
- The Huntington Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993; 72: 971-983.
- Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989; 245: 1066–1073.
- Corboy MJ, Thomas PJ, Wigley WC. CFTR Degradation and Aggregation Cystic Fibrosis Methods and Protocols. V70 of the series Methods in Molecular Medicine™. 2002; 277-294.
- Amm I, Sommer T, Wolf D. Protein quality control and elimination of protein waste the role of the ubiquitin proteasome system. BBA Mol Cell Res. 2014; 1843: 182–196.
- Ding WX, Yin XM. Sorting recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008; 4: 141–150.
- Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011; 10: 83–98.
- Sathasivam K, Hobbs C, Turmaine M, et al. Formation of polyglutamine inclusions in non CNS tissue. Hum Mol Genet. 1999; 8: 813–822.
- Yefimova MG, Béré E, Cantereau-Becq A, et al. Myelinosomes act as natural secretory organelles in Sertoli cells to prevent accumulation of aggregate prone mutant Huntingtin and CFTR. Hum Mol Genet. 2016 Aug 4. pii: ddw251. Hum Mol Genet. 2016 Oct 1;25(19):4170-4185. doi: 10.1093/hmg/ddw251.
- Blott EJ, Griffiths GM. Secretory lysosomes. Nat Rev Mol Cell Biol. 2002; 2: 122-131.
- Burgoyne RD, Morgan A. Secretory granule exocytosis. Physiol Rev. 2003; 83: 581-632.
- Huizing M, Helip Wooley A, Westbroek W, et al. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum. Genet. 2008; 9: 359-386.
- Marks MS, Heijnen HF, Raposo G. Lysosome-related organelles: unusual compartments become mainstream. Curr Opin Cell Biol. 2013; 25:495-505.
- Neves JS, Weller PF. Functional extracellular eosinophil granules: novel implications in eosinophil immunobiology. Curr Opin Immunol. 2009; 21: 694–699.
- Whitsett JA, Weaver TE. Hydrophobic surfactant proteins in lung function and disease. N Engl J Med. 2002; 347: 2141-2148.
- Piper RC, Katzmann DJ. Biogenesis and Function of Multivesicular Bodies Annu Rev Cell De. Biol. 2007; 23: 519–547.
- Jégou B. The Sertoli-germ cell communication network in mammals. Int Rev Cytol. 1993; 147: 25-96.
- Yefimova MG, Sow A, Fontaine I, et al. Dimeric transferrin inhibits phagocytosis of residual bodies by testicular rat Sertoli cells. Biol Reprod. 2008; 78: 697-704.
- Yefimova MG, Messaddeq N, Harnois T, et al. A chimerical phagocytosis model reveals the recruitment by Sertoli cells of autophagy for the degradation of ingested illegitimate substrates. Autophagy. 2013; 9: 653-666.
- Korah N, Smith CE, D'Azzo A, et al. Increase in macrophages in the testis of cathepsin a deficient mice suggests an important role for these cells in the interstitial space of this tissue. Mol Reprod Dev. 2003; 64:302-320.
- Yildiz Y, Matern H, Thompson B, et al. Mutation of beta glucosidase 2 causes glycolipid storage disease and impaired male fertility. J Clin Invest. 2006; 116: 2985-2994.
- Roy A, Lin Y-N, Matzuk MM. Shaping the sperm head: an ER enzyme leaves its mark. J Clin Invest. 2006; 116: 2860-2863.
- Chemes H. The phagocytic function of Sertoli cells a morphological, biochemical and endocrinological study of lysosomes and acid phosphatase localization in the rat testis. Endocrinology. 1986; 119: 1673-1681.