
PEDF: An essential stem cell regulator in models of Osteogenesis Imperfecta Type VI
Vikram Lyall1, Vivian Shih2*, and Chuhan Chung1,3*
1Department of Medicine, Yale University School of Medicine, Connecticut, USA
2Department of Orthopedics & Rehabilitation, Yale University School of Medicine, Connecticut, USA
3VA Connecticut Healthcare System, West Haven, CT, USA
Abstract
The rare bone disease Osteogenesis Imperfecta (OI) type VI is caused by mutations in the gene coding for PEDF, Serpinf1. Individuals with OI type VI have an accumulation of unmineralized bone matrix and multiple fractures. Our lab group has previously shown that PEDF restoration in the mouse model of OI type VI increases bone mass and mineralization. Further, we demonstrated that PEDF directs mesenchymal stem cells to the osteoblast lineage. One mechanism appears to involve modulation of canonical Wnt/β-catenin signaling in a temporally-defined manner. We have also induced pluripotent stem cells from a patient with OI type VI and differentiated the cells into osteoblasts to investigate how PEDF regulates the expression of various bone matrix proteins such as IBSP. In this brief review, we provide an overview of PEDF biology and highlight how PEDF’s role as a stem cell regulator lends support to its causative role in OI type VI.
PEDF Null Mutations Result in Osteogenesis Imperfecta Type VI
Osteogenesis Imperfecta (OI) type VI is an autosomal recessive bone disease characterized by decreased bone mineralization and multiple early fractures1,2. The histological markers of OI type VI are accumulations of unmineralized bone matrix (osteoid) and the appearance of a fish like pattern under polarized light1.
Unlike classic forms of OI that involve collagen mutations, OI type VI occurs in the setting of normal collagen synthesis, post-translations modification, and secretion1. Exome sequencing has identified null mutations in the gene coding for pigment epithelium-derived factor (PEDF), Serpinf1, as the causative genetic defect in OI type VI3. This results in complete absence of circulating PEDF4-6. The PEDF knockout (KO) mouse recapitulates key features of human OI type VI7. Another OI subtype, Type V, is caused by mutations in the gene coding for the bone enriched protein BRIL, IFITM58. In some instances, OI Type V presents with a phenotype consistent with the PEDF null state. Farber et al. have identified an IFITM5 mutation at codon 40 as the cause of this form of OI Type V. This mutation results in a S40L substitution and results in the functional absence of secreted PEDF8. How BRIL influences PEDF secretion remains unknown.
In addition to null mutations, homozygous in-frame insertion and deletion mutations in Serpinf1 have been identified and result in retention or degradation of PEDF within intracellular compartments. In these cases, PEDF levels are >10 fold lower compared to normal individuals and present clinically with a phenotype similar to PEDF null patients9. Recently, a point mutation in intron 6 of Serpinf1 (c.787-10C>G) has been identified as another genetic defect leading to OI type VI. This point mutation results in an altered splice site, addition of three amino acids (p.Lys262_Ile263insLeuSerGln), and an inability to secrete PEDF10. The absence of PEDF through null mutations or those that lead to its inability to be secreted result in OI type VI and provides insight into PEDF’s role as a key regulator of bone development and stem cell biology.
PEDF Biology
PEDF is a 50kDa secreted glycoprotein first identified and isolated from the conditioned medium of cultured human fetal retinal pigment epithelium cells11,12. PEDF is a member of the serine protease inhibitory (SERPIN) family, but lacks protease inhibitory function due to differences within the reactive center loop (RCL)12. Expression levels are highest in the liver and then adipose tissue13,14. Expression in other organs such as the eye, heart, pancreas, and skin suggests a broad distribution across many tissue types13. Serum PEDF is approximately 100nM in normal individuals, increased up to five-fold with obesity and the metabolic syndrome, and is undetectable or dramatically reduced in OI type VI15,16,6.
Prior to the discovery of the PEDF null state as the cause of OI type VI, many studies demonstrated PEDF’s role as a potent anti-angiogenic and neurotrophic factor17,18. Thus the recent identification of elevated PEDF as a biomarker for obesity and the metabolic syndrome has highlighted its diverse functions in many tissue types16,19,20. Sylvetsky et al. have shown that serum PEDF is sensitive to changes in body weight, raising the possibility that PEDF producing bone cells may couple changes in body weight to bone mass21.
The mechanisms by which PEDF plays a role in bone development remain incompletely understood. Initial work by Ma et al. identified PEDF’s interactions with the Wnt co-receptor low density lipoprotein receptor-related protein 6 (LRP6) and its inhibitory effects on the canonical Wnt pathway in retinal cells22. Since then, PEDF has been shown to inhibit Wnt signaling in the liver, pancreas, skin, and endothelial cells23-26. Due to the role of Wnt signaling in bone development and stem cell biology, our group and others have found that PEDF has complex effects on murine and human mesenchymal stem cells (MSCs)27. Exogenous PEDF appeared to stimulate the Wnt co-receptor LRP6 in early undifferentiated MSCs, while in later stages of terminal differentiation, PEDF appeared to block LRP6 activation. These effects on osteoblast lineage specification were accompanied by blockade of MSC to adipocyte differentiation that was restricted at an early time point. Once adipocyte lineage specification beyond 48 hours was achieved, PEDF could not block adipogenesis28. In cells without LRP6 PEDF mediated effects were not observed26. Further, our group found that PEDF KO mice displayed reduced bone volume and high fat content consistent with this effect on early MSC lineage specification27. These data provided in vivo evidence of PEDF’s role in modulating MSC differentiation to the osteoblast lineage while concurrently blocking adipocyte maturation.
PEDF’s Role in an Important Developmental Pathway
Other groups have shown that PEDF modulates MSC differentiation to the osteoblast lineage. Li et al. have shown that media supplemented with PEDF induces expression of osteoblast associated genes while also facilitating mineral deposition29. In addition, Niyibizi and colleagues demonstrated decreased osteoblast differentiation and mineralization in PEDF knockdown hMSC that were rescued with exogenous PEDF30. Together, these data provide evidence for PEDF’s direct role in osteoblast differentiation from MSC precursors and help explain why the absence of PEDF is the cause of a debilitating bone disease.
In our most recent work, we used the murine model of OI type VI (PEDF null state) to determine whether in vivo PEDF reconstitution would recover bone loss31. In mature mice (6 months old) PEDF injection had no effect in wild-type (WT) mice, but increased trabecular bone volume/total volume (BV/TV) by 52% in PEDF KO mice. Similarly, PEDF reconstitution recovered BV/TV by 35% in young PEDF KO mice (19 days old). Functional studies demonstrated that PEDF treatment improved various biomechanical parameters in PEDF KO femurs such as bone stiffness and plasticity. Notably, young mice had relatively less bone recovery compared to older mice, suggesting the possibility that older mice may have accumulated more unmineralized matrix characteristic of OI type VI.
To understand the mechanisms by which PEDF reconstitution recovers bone mass, we examined the canonical Wnt pathway in hMSCs undergoing osteoblast differentiation. A Wnt/β-catenin-GFP reporter demonstrated that Wnt activity was highest early in the differentiation time period but was absent by the end of the 21 day osteogenic differentiation protocol. Meanwhile, endogenously secreted PEDF increased over time, suggesting that it plays an important role in the terminal phase of osteoblast differentiation and directs MSCs to the osteoblast lineage in a temporally restricted manner. Continuous Wnt3a stimulation during the entire differentiation protocol resulted in decreased PEDF secretion and hMSC mineralization. However, mineralization was recovered when PEDF was added during the terminal phase of osteoblast differentiation. Therefore, our data suggest that PEDF antagonizes Wnt activity during osteoblast differentiation, thereby allowing for osteoblast maturation and matrix mineralization (Figure 1). To confirm PEDF’s inhibitory role of LRP6, we also showed that PEDF suppresses Wnt activity to the same extent as the well-known Wnt inhibitor, DKK1. In fact, DKK1 has also been shown to impede terminal osteoblast differentiation32, demonstrating that multiple endogenous inhibitors are present to turn off Wnt signaling. These mechanisms highlight the importance of PEDF-directed Wnt inhibition for proper osteoblast maturation.
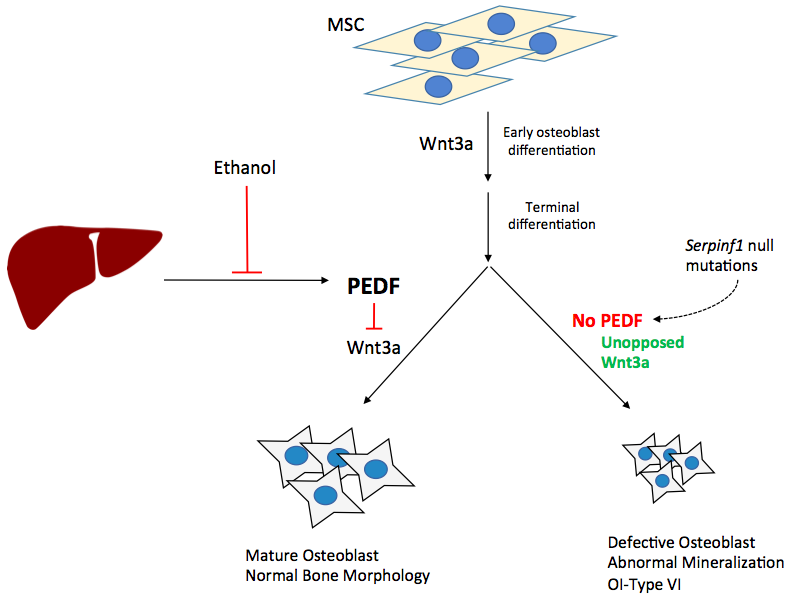
Figure 1. PEDF directed osteoblast differentiation:PEDF blocks Wnt3a ligand binding, allowing for proper osteoblast terminal differentiation. In the absence of PEDF, either in the PEDF null state (OI Type VI) or in the PEDF deficient state (alcohol induced PEDF suppression), continuous Wnt3a exposure hinders terminal osteoblast differentiation and normal mineralization. PEDF is necessary for osteoblast precursors to differentiate into mature osteoblasts. MSC, mesenchymal stem cell.
Generation of PEDF null iPSC
To assess the mechanisms leading to the OI type VI phenotype, we generated induced pluripotent stem cells (iPSCs) from a patient with OI type VI, differentiated them into MSCs, and assessed osteoblast-related gene expression33. Expression of bone sialoprotein, IBSP, was increased over 5-fold compared to control cells. IBSP is a major component of the extracellular matrix secreted by osteoblasts and its overproduction has been associated with reduced bone density and impaired bone homeostasis34,35. Interestingly, IBSP levels decreased in response to exogenous PEDF.
We have also seen that the PEDF 34mer peptide decreased levels of type I collagen in PEDF null iPSC (unpublished data). Thus, PEDF possesses multiple functions during bone development, including directing MSC differentiation and regulating extracellular matrix proteins.
In our cohort of hospitalizations for individuals with SCD from both California and Georgia, using Hb SS ICD-9-CM codes only (282.61, 282.62) versus any SCD code to identify a pediatric patient with SCA improved the positive predictive value (PPV) from 84% (9,850/11,740) to 93% (7,750/8,306); however, in the adult population the PPV only slightly improved from 79% (12,229/15,435) to 81% (10,352/12,752) (Table 2).
PEDF in Alcohol-Induced Bone Loss
Because the PEDF null state causes OI type VI, we questioned whether suppressed PEDF occurs in other disease states associated with bone loss. It is well established that alcohol abuse is a risk factor for osteoporosis and thus bone fragility36. In addition, alcoholic liver disease is associated with increased fractures37. The recent identification of secreted factors from the liver regulating bone mass support these clinical associations38. Given that PEDF is most highly expressed in the liver14, our most recent data is evaluating PEDF within the context of alcohol-induced bone loss. In wild-type and PEDF KO mice, ethanol feeding (5% Lieber-DeCarli diet) resulted in loss of trabecular bone volume in the femur and vertebral bodies. Bone mass was recovered following PEDF reconstitution in PEDF KO vertebral bodies (unpublished data). Moreover, we demonstrated that PEDF is suppressed in the bone and sera of wild-type mice fed an ethanol diet. This data is consistent with previous reports from our group demonstrating that loss of PEDF occurs in human and murine hepatic tissues with ethanol-induced steatosis39. This model highlights the role of PEDF in bone homeostasis and suggests that alcohol abuse may share similar mechanisms to the pathogenesis of OI type VI.
Conclusion
Initial work characterizing PEDF suggest that it is a critical factor modulating developmental pathways and stem cells. First studied as a neuronal differentiation factor in retinoblastoma cells, in vivo studies demonstrated that PEDF induced neuroblastoma cells into a terminally differentiated state12,40-42. However, PEDF in vivo transplantation in retinoblastoma cells resulted in tumor growth, suggesting that PEDF effects are context specific and in stem cell populations can lead to either cell renewal or differentiation43,44. PEDF in vitro strongly affected the differentiation of photoreceptors and counteracted gliotoxic damage, i.e. inflammation, of radial glial cells45,46.
While this paradox is still incompletely understood, the discoveries of the PEDF null state in OI Type VI and PEDF’s role as a Wnt agonist in many tissue sites have explained its diverse functionality. As a fundamental developmental pathway, Wnt signaling is crucial in development and adult homeostasis. As a result, aberrant Wnt signaling is associated with a wide array of abnormalities including various bone diseases, diabetes, and irregular angiogenesis in diabetic retinopathy47-49. Thus, it is important to determine if PEDF-directed Wnt signaling occurs in disease states where Wnt signaling is disrupted. In addition, it would be useful to understand how Wnt signaling is altered in PEDF deficient disease states such as alcoholic osteodystrophy.
Grant support: This work was supported by Digestive Diseases Research Core Center–5P30DK034989 (CC); NIH/NIAAA R2123607; Veterans Affairs Merit Grant (CC).
References
- Glorieux FH, Ward LM, Rauch F, et al. Osteogenesis imperfecta type VI: A form of brittle bone disease with a mineralization defect. J Bone Miner Res. 2002; 17: 30– 38.
- Roughley PJ, Rauch F, Glorieux FH. Osteogenesis imperfect Clinical and molecular diversity. Eur Cells Mater. 2003; 5: 41–47.
- Becker J, Semler O, Gilissen C, et al. Exome sequencing identifies truncating mutations in human serpinf1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2011; 88: 362–371.
- Homan EP, Rauch F, Grafe I, et al. Mutations in SERPINF1 cause Osteogenesis imper- fecta type VI. J Bone Miner Res 2011; 26: 2798-803.
- Venturi G, Gandini A, Monti E, et al. Lack of expression of SERPINF1, the gene coding for pigment epithelium-derived factor, causes progressively deforming Osteogenesis imperfecta with normal type I collagen. J Bone Miner Res. 2012; 27: 723-8.
- Rauch F, Husseini A, Roughley P, et al. Lack of circulating pigment epithelium-derived factor is a marker of Osteogenesis imperfecta type VI. J Clin Endocrinol Metab. 2012; 97: E1550-6.
- Bogan R, Riddle RC, Li Z, et al. A mouse model for human osteogenesis imperfecta type VI. J Bone Miner Res. 2013; 28: 1531–1536.
- Farber CR, Reich A, Barnes AM, et al. A novel IFITM5 mutation in severe atypical Osteogenesis imperfecta type VI impairs osteoblast production of pigment epithelium-derived factor. J Bone Miner Res. 2014; 29: 1402-11.
- Al Jallad H, Palomo T, Roughley P, et al. The effect of SERPINF1 in- frame mutations in Osteogenesis imperfecta type VI. Bone. 2015; 76: 115-20.
- Ward, Leanne, Moffatt P. Osteogenesis imperfecta type VI in individuals from Northern Canada. Calcified tissue international. 2016 98.6: 566-572.
- Tombran Tink J, Johnson LV. Neuronal differentiation of retinoblastoma cells induced by medium conditioned by human RPE cells. Invest Ophthalmol Vis Sci. 1989; 30: 1700-7.
- FR, Chader GJ, Johnson LV, et al. Pigment epithelium-derived factor: neurotrophic activity and identification as a member of the serine protease inhibitor gene family. Proc Natl Acad Sci U S A. 1993; 90: 1526-30.
- Tombran Tink J, Mazuruk K, Rodriguez IR, et al. Organization, evolutionary conservation, expression and unusual Alu density of the human gene for pigment epithelium-derived factor, a unique neurotrophic serpin. Mol Vis. 1996; 2: 11.
- Uhlen M, Fagerberg L, Hallstrom BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347: 1260419.
- Petersen SV, Valnickova Z, Enghild JJ. Pigment-epithelium-derived factor (PEDF) occurs at a physiologically relevant concentration in human blood: purification and characterization. Biochem J. 2003; 374: 199-206.
- Yamagishi S, Adachi H, Abe A, et al. Elevated serum levels of pigment epithelium-derived factor in the metabolic syndrome. J Clin Endocrinol Metab. 2006; 91: 2447-50.
- Tombran Tink J, Barnstable CJ. PEDF: a multifaceted neurotrophic factor. Nat Rev Neurosci. 2003; 4: 628-36.
- Dawson DW, Volpert OV, Gillis P, et al. Pigment epithelium-derived factor a potent inhibitor of angiogenesis. Science. 1999; 285: 245-8.
- Chen C, Tso AW, Law LS, et al. Plasma level of pigment epithelium-derived factor is independently associated with the development of the metabolic syndrome in Chinese men: a 10-year prospective study. J Clin Endocrinol Metab. 2010; 95: 5074-81.
- Choi KM, Hwang SY, Hong HC, et al. C1q/TNF-related protein-3 (CTRP-3) and pigment epithelium-derived factor (PEDF) concentrations in patients with type 2 diabetes and metabolic syndrome. Diabetes. 2012; 61: 2932-6.
- Sylvetsky, Allison C, Chandran A. Pigment Epithelium-Derived Factor Declines in Response to an Oral Glucose Load and Is Correlated with Vitamin D and BMI but Not Diabetes Status in Children and Young Adults. Hormone Research in Paediatrics. 2017; 87.5: 301-306.
- Park K, Lee K, Zhang B, et al. Identification of a novel inhibitor of the canonical Wnt pathway. Mol Cell Biol. 2011; 31: 3038-51.
- Protiva P, Gong J, Sreekumar B, et al. Pigment Epithelium-Derived Factor (PEDF) inhibits Wnt/-catenin signaling in the liver. Cell Mol Gastroen terol Hepatol. 2015; 1: 535-49.
- Gong J, Belinsky G, Sagheer U, et al. Pigment Epithelium-derived Factor (PEDF) Blocks Wnt3a Protein-induced Autophagy in Pancreatic Intraepithelial Neoplasms. J Biol Chem. 2016; 291: 22074-22085.
- Qi W, Yang C, Dai Z, et al. High levels of pigment epithelium-derived factor in diabetes impair wound healing through suppression of Wnt signaling. Diabetes. 2014; 64: 1407–1419.
- Ma S, Yao S, Tian H, et al. Pigment epithelium-derived factor alleviates endothelial injury by inhibiting Wnt/β-catenin pathway. Lipids in Health and Disease. 2017; 16(1): 31.
- Gattu AK, Swenson ES, Iwakiri Y, et al. Determination of mesenchymal stem cell fate by pigment epithelium- derived factor (PEDF) results in increased adiposity and reduced bone mineral content. FASEB J. 2013; 27: 4384–4394.
- Wang, Min, Park K. Pigment epithelium-derived factor suppresses adipogenesis via inhibition of the MAPK/ERK pathway in 3T3-L1 preadipocytes. Am J Physiology-Endocrinology and Metabolism. 2009; 2976: E1378-E1387.
- Li F, Song N, Tombran Tink J, et al. Pigment epithelium-derived factor enhances differentiation and mineral deposition of human mesenchymal stem cells. Stem Cells. 2013; 31: 2714–2723.
- Li F, Song N, Tombran Tink J, et al. Pigment epithelium derived factor suppresses expression of sost/sclerostin by osteocytes: implication for its role in bone matrix mineralization. J Cell Physiol. 2015; 230: 1243-9.
- Belinsky GS, Sreekumar B, Andrejecsk JW, et al. Pigment epithelium–derived factor restoration increases bone mass and improves bone plasticity in a model of osteogenesis imperfecta type VI via Wnt3a blockade. The FASEB Journal. 2016; 30(8): 2837-48.
- van der Horst G, van der Werf SM, Farih Sips H, et al. Downregulation of Wnt signaling by increased expression of Dickkopf-1 and -2 is a prerequisite for late-stage osteoblast differentiation of KS483 cells. J Bone Miner Res. 2005; 20: 1867-77.
- Belinsky GS, Ward L, Chung C. Pigment epithelium-derived factor (PEDF) normalizes matrix defects in iPSCs derived from Osteogenesis imperfecta Type VI. Rare Diseases. 2016; 4(1): e1212150.
- Valverde P, Zhang J, Fix A, et al. Overexpression of bone sialoprotein leads to an uncoupling of bone formation and bone resorption in mice. J Bone Miner Res. 2008; 23: 1775-88.
- Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, et al. New sequence variants associated with bone mineral density. Nat Genet. 2009; 41: 15-7.
- Kanis JA, Borgstrom F, De Laet C, et al. Assessment of fracture risk. Osteoporos Int. 2005; 16: 581–589.
- Bang CS, Shin IS, Lee SW, et al. Osteoporosis and bone fractures in alcoholic liver disease: A meta-analysis. World Journal of Gastroenterology: 2015; WJG 21(13): 4038.
- Chung, Chuhan, Karl L. Insogna. "The liver throws the skeleton a bone (resorption factor)." Hepatology. 2016; 64.3: 977.
- Chung C, Shugrue C, Nagar A, et al. Ethanol exposure depletes hepatic pigment epithelium-derived factor, a novel lipid regulator. Gastroenterology. 2009; 136: 331-340 e332.
- Becerra SP, Palmer I, Kumar A, et al. Overexpression of fetal human pigment epithelium-derived factor in Escherichia coli. A functionally active neurotrophic factor. J Biol Chem. 1993; 268: 23148-56.
- Tombran Tink J, Chader GG, Johnson LV. PEDF: a pig- ment epithelium-derived factor with potent neuronal dif- ferentiative activity. Exp Eye Res. 1991; 53: 411-4.
- Crawford SE, Stellmach V, Ranalli M, et al. Pigment epithelium-derived factor (PEDF) in neuroblastoma: a multifunctional mediator of Schwann cell antitumor activity. J Cell Sci. 2001; 114: 4421-8.
- Seigel GM, Tombran Tink J, Becerra SP, et al. Differentiation of Y79 retinoblastoma cells with pigment epithelial-derived factor and interphotoreceptor matrix wash effects on tumorigenicity. Growth Factors. 1994; 10: 289-97.
- Ramirez Castillejo C, Sanchez Sanchez F, Andreu Agullo C,et al. Pigment epithelium-derived factor is a niche signal for neural stem cell renewal. Nat Neurosci. 2006; 9: 331-9.
- Thangaraj G, Christophel J, Bachmann G, et al. PEDF counteracts DL-α-aminoadipate toxicity and rescues gliotoxic damages in RPE-free chicken retinal explants. Experimental eye research. 2015; 134: 111-22.
- Volpert KN, Tombran Tink J, Barnstable C, et al. PEDF and GDNF are key regulators of photoreceptor development and retinal neurogenesis in reaggregates from chick embryonic retina. Journal of ocular biology, diseases, and informatics. 2009 ; 2(1): 1-1.
- Liu H, Fergusson MM, Wu JJ, et al. Wnt signaling regulates hepatic metabolism. Sci Signal. 2011; 4: ra6.
- Liu W, Singh R, Choi CS, et al. Low density lipoprotein (LDL) receptor-related protein 6 (LRP6) regulates body fat and glucose homeostasis by modulating nutrient sensing pathways and mitochon- drial energy expenditure. J Biol Chem. 2012; 287: 7213- 23.
- Dejana E. The role of wnt signaling in physiological and pathological angiogenesis. Circ Res. 2010; 107: 943-52.