
Diagnosing and treatment of Fabry's disease from a neurologic perspective
Michael Y. Soliman, Rima El-Abassi* and John D. England
Department of Neurology, South East Louisiana Veteran Health Care Sysytem (SELVHCS), Louisiana State University, New Orleans, Louisiana, 70112, United States
Introduction
Despite the surge in our knowledge of Fabry’s Disease (FD) and the advancements in diagnostic tools, there is often a significant time between the onset of symptoms and diagnosis. According to the Fabry Registry, 10.5 years is the average age of symptom onset, but 28.5 years is the average age of diagnosis. As a rare disease which shares clinical manifestations with many other disorders, FD can be challenging to recognize. This is a concern because early initiation of enzyme replacement therapy has been found to be important for reduction of disease burden. In order to help clinicians identify FD in its earliest stages, in this review we describe its neurological features, diagnosis and treatment.
Characteristic features of neuropathy in Fabry’s disease:
Neuropathy in FD is a small fiber neuropathy (SFN), affecting small myelinated and unmyelinated neurons. The characteristic neuropathy of FD is a symmetrical, length-dependent sensory polyneuropathy which starts in the feet and spreads proximally. In FD the Aδ fibers are preferentially affected, which is in contrast to most other SFNs where both Aδ- and C-fibers are affected1.
Clinical presentation
Symptoms usually begin between the ages of 3 to 10 years, with males affected earlier and more severely than females2-4. FD is divided into “classical” and “non-classical” or “atypical” varieties. Burning pain, hypo or hyperhidrosis, transient ischemic attacks, strokes, angiokeratoma, proteinuria, cardiomyopathy, arrhythmia, cochleo-vestibular and gastrointestinal disorders are the most common presenting features in “classically” affected hemizygous males who have no residual α-galactosidase A activity4. Patients with “non-classical” or “atypical” disease who have residual α-galactosidase A activity exhibit milder disease, and typically present in the fourth to sixth decades with clinical manifestations confined to one organ system5,6.
Pain in FD
Pain is experienced by 60–80% of affected males and females FD patients7,8 with SFN. Two types of pain associated with SFN have been described. The first type is the episodic painful crisis (Fabry's crisis), which is characterized by agonizing burning pain starting in the extremities and radiating centripetally. It may be precipitated by fever, exercise, fatigue, stress or rapid changes in temperature9. The second type is chronic pain, which is characterized by burning, shooting pain or dysesthesias in the hands or feet9,10. Decrease in pain with ageing occurs in some patients11. This may occur as a consequence of progressive small fiber damage with changes in neural function leading to a decrease in pain12. Therefore when pain is not an active symptom, it is important to inquire about a history of acroparesthesias in childhood.
Other sensory manifestations
There is loss of temperature sensation in the hands and feet and reduced tolerance to cold13.
Autonomic nervous system involvement
There is controversy whether autonomic neuropathy is a major feature of FD. Since FD causes relatively selective damage to Aδ fibers, autonomic functions are usually preserved14. Sweat glands14–19, dermal nerve endings9, vessel response20, heart rate variability21, orthostatic blood pressure, and male sexual function are usually normal22. These observations suggest that it is unlikely that FD patients suffer significant autonomic neuropathy. A Four-year prospective clinical trial found remarkable improvement in all heart rate variability indices in boys undergoing agalsidase alfa replacement therapy23.There are a few reports of orthostatic hypotension and syncope in patients with FD, suggesting localized cardiovascular autonomic abnormalities24,25. However, these and other abnormal studies of peripheral hemodynamics might be explained by end-organ failure such as stiffness of vascular smooth muscle and endothelial dysfunction26.
Cerebrovascular disease
In FD there is a significant incidence of stroke, which is mostly small vessel ischemic event27, and there is also a predilection for acute ischemia in the posterior cerebral circulation28,29. Among different MRI changes, the finding of hyper-intensity in the pulvinar on T1 weighted images (“pulvinar sign”) has been found to be a highly specific sign in FD30 (Fig. 1). The most common angiographic findings are tortuous, elongated, ectatic vertebral and basilar arteries. Increased basilar artery diameter is 87% accurate in diagnosing FD31. Cerebral blood flow changes may result in increased white matter interstitial pressure and metabolic impairment32. Cerebral involvement is usually widespread. The posterior cerebrum is predominantly involved32. Chiari type I malformation has been found in some patients with FD33.
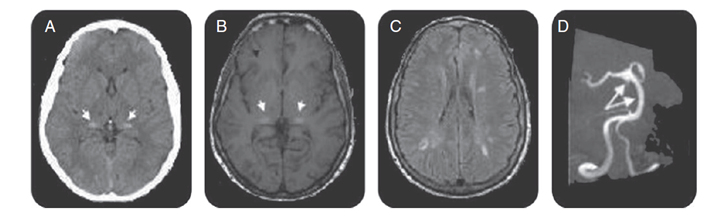
Figure 1: Brain CT, brain MRI, and magnetic resonance angiography for FD patients.
R. El-Abassi et al. / Journal of the Neurological Sciences 344 (2014) 5–19
Axial CT scan demonstrated increased attenuation in the pulvinar region (arrows, A), corresponding to the sites of hyper-intensity on T1-weighted MRI (the pulvinar sign) (arrows, B). Fluid-attenuated inversion recovery–weighted axial MRI section showed multiple white matter lesions in cerebral hemispheres (C). Magnetic resonance angiography shows basilar dolichoectasia (arrows, D).
Hearing loss
There is a high incidence of progressive hearing loss, mainly sensorineural with vestibular dysfunction and sudden deafness in male patients with FD34.
Diagnosis
When to suspect Fabry’s disease
To diagnose FD, further specific tests are recommended by the National Society of Genetic Counselors for patients with any of the factors outlined in Table 1.
| Test any patient who has | In the absence of factors 1 and 2, test patients with at least two of the features below |
|---|---|
| A family history of Fabry Disease | Decreased sweating (anhidrosis or hypohidrosis) |
| OR | Reddish-purple skin rash in the bathing trunk area (angiokeratomas) |
| Corneal verticillata (“whorls”) on slit lamp exam | Personal and/or family history of kidney failure |
| Personal or family history of “burning” or “hot” pain in the hands and feet, particularly during fevers (acroparesthesias) | |
| Personal or family history of exercise, heat, or cold intolerance. | |
| Patients with sporadic or non-autosomal dominant (no male-to-male) transmission of unexplained cardiac hypertrophy |
Table 135National Society of Genetic Counselors recommendations for testing patients for FD
Diagnostic testing
In males, the diagnosis is based on typical FD signs and symptoms, very low or completely absent α-galactosidase A (GLA) activity in leukocytes, plasma or fibroblasts, increased globotriaosylceramide (Gb3) and lyso-Gb3 concentrations in plasma and urine and a pathogenic mutation on genetic analysis29,36,37 (Figure 2)1. Females affected by FD have α-galactosidase A levels which can range from deficient to normal3,38-40. Therefore, diagnosis must rely upon molecular analysis to identify a disease causing mutation in the GLA gene2,41 (Figure 3)1. Prenatal diagnosis is feasible by determining the α-galactosidase A activity in cultured chorionic villi at 10 weeks of pregnancy or in cultured amniotic cells at 14 weeks of pregnancy42. Neonatal screening for FD in males is technically feasible by measurement of α-galactosidase A activity in dried blood spot (DBS) using either the fluorogenic or mass spectrometric substrate and will detect cases both with complete deficiency and residual enzyme activity43.
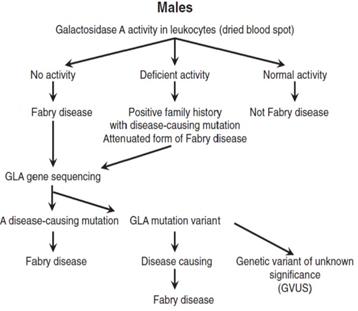
Figure 2: Proposed algorithm for testing males with suspected Fabry Disease
R. El-Abassi et al. / Journal of the Neurological Sciences 344 (2014) 5–19
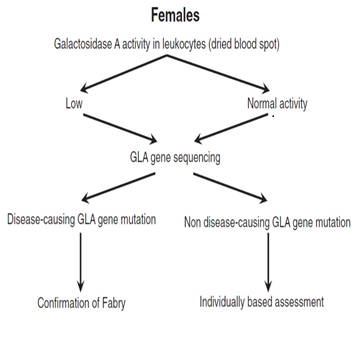
Figure 3: Proposed algorithm for testing females with suspected Fabry Disease
R. El-Abassi et al. / Journal of the Neurological Sciences 344 (2014) 5–19
Neurologic testing
Conventional nerve conduction studies (NCS) assess only large myelinated nerve fibers, and are usually normal in FD unless renal failure is present. Quantitative sensory testing (QST), quantitative sudomotor axon reflex test (QSART) and skin biopsy to assess epidermal nerve fiber density (ENFD) are useful to confirm the presence of SFN. Recently, pain-related evoked potentials (PREPs), where electrical current using special concentric electrodes44 is used to stimulate A-delta fibers, was described as a suitable and easily applicable new tool for objective small fiber diagnostics45. SFN in FD has been demonstrated to be length dependent by using proximal and distal skin biopsy sites46. Corneal nerve fiber density (C-fibers) and corneal sensation has been demonstrated to be significantly reduced47. Some studies concluded that sensory impairment and SF pathology in FD patients are gender-dependent, associated with reduced renal function, and are progressive in most patients despite ERT48,49.
Differential diagnosis
For clinicians, it is of utmost importance to distinguishing FD from other causes of painful small-fiber neuropathy (Table 2)1.
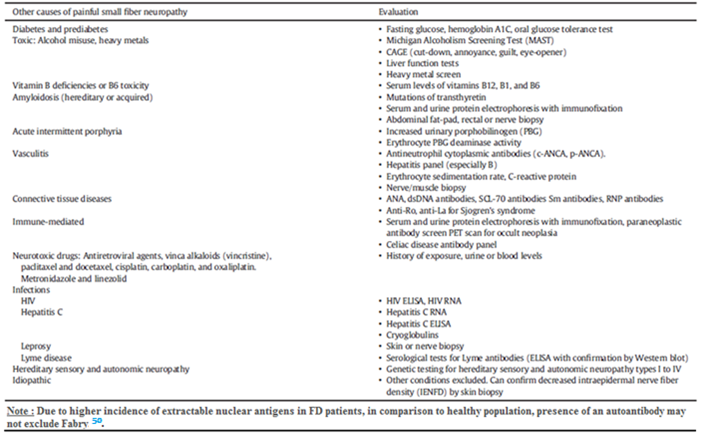
Table 2
Differential diagnosis of small fiber painful neuropathy
R. El-Abassi et al. / Journal of the Neurological Sciences 344 (2014) 5–19
Management
Enzyme replacement therapy (ERT)
ERT is currently the standard of care for FD patients. The most current literature emphasizes that ERT should be instituted upon confirming the diagnosis of FD. Recombinant human α-galactosidase A is commercially available in two forms given as an intravenous (IV) infusion every two weeks. Agalsidase alfa (Replagal) and Agalsidase beta (Fabrazyme) are the two commercially available preparations. Agalsidase beta is approved for use in the United States (US). Agalsidase alfa, on the other hand, is not approved for use in the US. Although complications such as stroke and renal damage may still occur while receiving ERT, patients with FD were found to benefit from ERT, particularly when started at earlier stages of the disease before end organ damage occurs51.
Infusion related reactions and neutralizing IgG antibodies with ERT
Adverse effects of ERT include fever, chills, facial flushing and rigors. Infusion reactions are managed conservatively with antihistamines and corticosteroids in cases of a severe reaction. Neutralizing IgG antibodies may develop to agalsidase alfa and beta and show cross reactivity between the two enzyme replacements52–54. It is recommended that patients receiving ERT should be checked for serum neutralizing antibodies every three months during the first year and then yearly55.
Pain management
Anti-epileptic drugs are widely used for pain control in patients with FD. Treatment using combinations of anti-epileptic drugs may be necessary for adequate pain control56. Treatment with agalsidase beta resulted in decreased neuropathic pain and improved function of Aδ, C and Aβ nerve fibers and intradermal vibratory receptors detected by QST57,58. Analgesic medications can also be used, but nonsteroidal anti-inflammatory drugs (NSAIDs) should be avoided since they are not effective and potentially harmful to kidneys59. In an effort to provide practical pain management guidelines for FD in adults, an FD Expert Panel suggested recommendations that are summarised in Table.360.
| Indication | Agent | Type of pain |
|---|---|---|
| First-line | • Tricyclic antidepressants | |
| • Serotonin and norepinephrine reuptake inhibitors (e.g., duloxetine, venlafaxine) | chronic neuropathic pain | |
| • Carbamazepine | ||
| • Gabapentin | ||
| • Gabapentinoid pregabalin | ||
| Second-line | • Lidocaine patches | |
| • Topical capsaicin (8%) patches | ||
| • Tramadol | acute pain | |
| Third-line | • Strong opioids | acute pain |
| • Cannabinoids | ||
| Fourth-line | • Methadone | |
| • Anticonvulsants with lesser evidence of efficacy (e.g., lamotrigine, lacosamide) | ||
| • Tapentadol | ||
| • Botulinum toxin. | ||
| Other agents to consider | ||
| Acute-pain prophylaxis before physical activity | • Lidocaine or capsaicin cream | |
| During pain crises | • Phenytoin | acute pain chronic neuropathic pain |
| • Intravenous lidocaine | acute pain | |
| Nonsteroidal anti-Inflammatory drugs | • Ibuprofen | acute pain |
| • Diclofenac | ||
| Therapy-resistant neuropathic pain |
• Opioid • Ketamine |
|
| Note: These recommendations are based on an FD Expert Panel that convened in Rome, Italy, in March 2014 and on subsequent discussions. | ||
Table 3Recent recommendations for pain treatment in Fabry`s disease60
Peripheral nervous system
Agalsidase alfa therapy has been shown to achieve lower heat pain thresholds, improved cold detection threshold, and improvement of sympathetic skin responses with associated improvement in acroparesthesia and anhidrosis57,58,61,62.. Treatment with agalsidase beta resulted in improved function of Aδ, C and Aβ nerve fibers and intradermal vibratory receptors detected by QST57,58.
Cerebrovascular system
Neither agalsidase alfa nor beta has been shown to reduce the frequency of cerebrovascular events63. Possible reasons are that neither of the available enzyme compounds cross the blood brain barrier and that irreversible damage to the endothelium typically occurs before the initiation of therapy. Thus, other preventive measures including the administration of antithrombotic agents and control of risk factors are indicated.
Pharmacological chaperone therapy
Migalastat was recently approved by the European commission for treatment of FD patients aged 16 years and older, but not yet approved in the US. It works by facilitating appropriate trafficking of GLA to lysosomes. Migalastat is orally administered, thus it is less invasive than ERT IV infusion. Reduction in plasma lyso-Gb3 levels and improvement in kidneys, heart and gastrointestinal tract functions, have been reported in patients with suitable mutations. No sufficient evidence was found for the effect of Migalastat on neurological manifestations of FD64.
Conclusion
FD is rare and shows diverse symptoms in the initial stages of manifestation; therefore, the definitive diagnosis is often delayed. However, in recent years, FD has become more widely recognized and there has been a considerable advance in the diagnosis and treatment options. The response to ERT is dependent on the severity of organ involvement, and better results occur when the treatment is instituted early in the disease before major organ damage has occurred.
Conflict of interest
The author certifies that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
References
- El Abassi R, Singhal D, England JD. Fabry’s disease. J Neurol Sci. 2014; 344: 5–19.
- Sakuraba H, Oshima A, Fukuhara Y, et al. Identification of point mutations in the alpha galactosidase A gene in classical and atypical hemizygotes with Fabry disease. Am J Hum Genet. 1990; 47(5): 784–9 (Epub 1990/11/01).
- WilcoxWR, Oliveira JP, Hopkin RJ, et al. Females with Fabry disease frequently have major organ involvement lessons from the Fabry registry. Mol Genet Metab. 2008; 93(2): 112–28 (Epub 2007/11/27).
- Mehta A, Ricci R, Widmer U, et al. Fabry disease defined baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Investig. 2004; 34(3): 236–42.
- von Scheidt W, Eng CM, Fitzmaurice TF, et al. An atypical variant of Fabry's disease with manifestations confined to the myocardium. N Engl J Med. 1991; 324(6): 395–9 (Epub 1991/02/07).
- Nakao S, Takenaka T, Maeda M, et al. An atypical variant of Fabry's disease in men with left ventricular hypertrophy. N Engl J Med. 1995; 333(5): 288–93 (Epub 1995/08/03).
- Hopkin RJ, Bissler J, Banikazemi M, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry registry. Pediatr Res. 2008; 64(5): 550–5 (Epub 2008/07/04).
- Hoffmann B, Beck M, Sunder-Plassmann G, et al. Nature and prevalence of pain in Fabry disease and its response to enzyme replacement therapy a retrospective analysis from the Fabry Outcome Survey. Clin J Pain. 2007; 23(6): 535–42 (Epub 2007/06/19).
- Charrow J. A 14 year old boy with pain in hands and feet. Pediatr Ann. 2009; 38(4): 190 (2. Epub 2009/05/22).
- Schiffmann R, Scott LJ. Pathophysiology and assessment of neuropathic pain in Fabry disease. Acta Paediatr Suppl. 2002; 91(439): 48–52 (Epub 2003/02/08).
- Kolodny EH, Pastores GM. Anderson Fabry disease extrarenal neurologic manifestations. J Am Soc Nephrol. 2002; 13: S3–S150.
- Rahman AN, Simeone FA, Hackel DB, et al. Angiokeratoma corporis diffusum universale hereditary dystopic lipidosis. Trans Assoc Am Phys. 1971; 74: 366–77.
- Hilz MJ, Stemper B, Kolodny EH. Lower limb cold exposure induces pain and prolonged small fiber dysfunction in Fabry patients. Pain. 2000; 84(2–3): 361–5 (Epub 2000/02/10).
- Donaghy M, Hakin RN, Bamford JM, et al. Hereditary sensory neuropathy with neurotrophic keratitis Description of an autosomal recessive disorder with a selective reduction of small myelinated nerve fibres and a discussion of the classification of the hereditary sensory neuropathies. Brain. 1987; 110(Pt 3): 563–83 (Epub 1987/06/01).
- Cable WJ, Kolodny EH, Adams RD. Fabry disease: impaired autonomic function. Neurology. 1982; 32(5): 498–502 (Epub 1982/05/01).
- Fukuhara N, Suzuki M, Fujita N, et al. Fabry's disease on the mechanism of the peripheral nerve involvement. Acta Neuropathol. 1975; 33(1): 9–21 (Epub 1975/10/27).
- Yamamoto K, Sobue G, Iwase S, et al. Possible mechanism of anhidrosis in a symptomatic female carrier of Fabry's disease an assessment by skin sympathetic nerve activity and sympathetic skin response. Clin Auton Res. 1996; 6(2): 107–10 (Epub 1996/04/01).
- Lao LM, Kumakiri M, Mima H, et al. The ultrastructural characteristics of eccrine sweat glands in a Fabry disease patient with hypohidrosis. J Dermatol Sci. 1998; 18(2): 109–17 (Epub 1998/12/02).
- Schiffmann R, Kopp JB, Austin III HA, et al. Enzyme replacement therapy in Fabry disease a randomized controlled trial. JAMA. 2001; 285(21): 2743–9 (Epub 2001/06/21).
- Altarescu G, Moore DF, Pursley R, et al. Enhanced endothelium dependent vasodilation in Fabry disease. Stroke. 2001; 32(7): 1559–62 (Epub 2001/07/07).
- Kampmann C, Wiethoff CM, Whybra C, et al. Cardiac manifestations of Anderson Fabry disease in children and adolescents. Acta Paediatr. 2008; 97(4): 463–9 (Epub 2008/03/28).
- Biegstraaten Marieke, van Schaik Ivo N, Wieling Wouter, et al. Autonomic neuropathy in Fabry disease a prospective study using the Autonomic Symptom Profile and cardiovascular autonomic function tests. BMC Neurol. 2010; 10: 38 (Epub 2001/12/12.).
- Schiffmann R, Martin RA, Reimschisel T, et al. Four year prospective clinical trial of agalsidase alfa in children with Fabry disease. J Pediatr. 2010; 156(5): 832-837 837 e831.
- Lien YH, Lai LW, Lui CY. Unexpected diagnosis of fabry disease in an 80 year old man with syncope. Cardiology. 2001; 96(2): 115–6 (Epub 2001/12/12).
- Mutoh T, Senda Y, Sugimura K, et al. Severe orthostatic hypotension in a female carrier of Fabry's disease. Arch Neurol. 1988; 45(4): 468–72 (Epub 1988/04/01).
- Biegstraaten M, Hollak CE, Bakkers M, et al. Small fiber neuropathy in Fabry disease. Mol Genet Metab. 2012; 106(2): 135–41 (Epub 2012/ 04/14).
- Sims K, Politei J, BanikazemiM, et al. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events natural history data from the Fabry registry. Stroke. 2009; 40(3): 788–94 (Epub 2009/01/20).
- Fazekas F, Enzinger C, Schmidt R, et al. MRI in acute cerebral ischemia of the young the Stroke in Young Fabry Patients sifap1 study. Neurology. 2013; 26(22): 1914–21 (81).
- Mitsias P, Levine SR. Cerebrovascular complications of Fabry's disease. Ann Neurol. 1996; 40(1): 8–17 (Epub 1996/07/01).
- Burlina AP, Manara R, Caillaud C, et al. The pulvinar sign frequency and clinical correlations in Fabry disease. J Neurol. 2008; 255(5): 738–44 (Epub 2008/02/26).
- Fellgiebel A, Keller I, Marin D, et al. Diagnostic utility of different MRI and MR angiography measures in Fabry disease. Neurology. 2009; 72(1): 63–8 (Epub 2009/01/06).
- Hilz MJ, Kolodny EH, Brys M, et al. Reduced cerebral blood flow velocity and impaired cerebral autoregulation in patients with Fabry disease. J Neurol. 2004; 251(5): 564–70 (Epub 2004/05/28).
- Germain DP, Benistan K, Halimi P. Chiari type I malformation in four unrelated patients affected with Fabry disease. Eur J Med Genet. 2006; 49(5): 419–25 (Epub 2006/03/03).
- Germain DP, Avan P, Chassaing A, et al. Patients affected with Fabry disease have an increased incidence of progressive hearing loss and sudden deafness: an investigation of twenty two hemizygous male patients. BMC Med Genet. 2002; 3: 10 (Epub 2002/10/16).
- Laney DA, Bennett RL, Clarke V, et al. Fabry disease practice guidelines Recommendations of the national society of genetic counselor. J Genet Counsel. 2013; 22: 555–64.
- Mayes JS, Scheerer JB, Sifers RN, et al. Differential assay for lysosomal alpha galactosidases in human tissues and its application to Fabry's disease. Clin Chim Acta. 1981; 112(2): 247–51 (Epub 1981/05/05).
- Hoffmann B, Georg Koch H, Schweitzer Krantz S, et al. Deficient alpha galactosidase A activity in plasma but no Fabry disease a pitfall in diagnosis. Clin Chem Lab Med. 2005; 43(11): 1276–7 (Epub 2005/10/20).
- MacDermot KD, Holmes A, Miners AH. Anderson Fabry disease clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001; 38(11): 769–75 (Epub 2001/12/06).
- Germain DP. Fabry disease Clinical and genetic aspects Therapeutic perspectives La Revue de medecine interne/fondee par la Societe nationale francaise de medecine interne. La maladie de Fabry Aspects cliniques et genetiques Perspectives therapeutiques.2000; 21(12): 1086-103. Epub 2001/02/24.
- Whybra C, Wendrich K, Ries M, et al. Clinical manifestation in female Fabry disease patients. Contrib Nephrol. 2001; 136: 245–50 (Epub 2001/11/02).
- Germain DP, Benistan K, Angelova L. X-linked inheritance and its implication in the diagnosis and management of female patients in Fabry disease. La Revue de medecine interne fondee par la Societe nationale francaise de medecine interne 31 (Suppl. 2). 2010; S209–13 (Epub 2011/01/08.168).
- Gal A, Hughes DA, Winchester B. Toward a consensus in the laboratory diagnostics of Fabry disease recommendations of a European expert group. J Inherit Metab Dis. 2011; 34(2): 509–14.
- Spada M, Pagliardini S, Yasuda M, et al. High incidence of later onset fabry disease revealed by newborn screening. Am J Hum Genet. 2006; 79: 31–40.
- Kaube H, Katsarava Z, Kaufer T, et al. A new method to increase nociception specificity of the human blink reflex. Clin Neurophysiol. 2000; 111: 413–6.
- Üçeyler N, Kahn AK, Kramer D, et al. Impaired small fiber conduction in patients with Fabry disease a neurophysiological case control study. BMC Neurol. 2013; 13: 47.
- Torvin Moller A, Winther Bach F, Feldt-Rasmussen U, et al. Functional and structural nerve fiber findings in heterozygote patients with Fabry disease. Pain. 2009; 145(1–2): 237–45 (Epub 2009/08/12).
- Liguori R, Di Stasi V, Bugiardini E, et al. Small fiber neuropathy in female patients with fabry disease. Muscle Nerve. 2010; 41(3): 409–12.
- Gemignani F, Marbini A, Bragaglia MM, et al. Pathological study of the sural nerve in Fabry's disease. Eur Neurol. 1984; 23: 173–81.
- Kennedy WR. Unmyelinated nerves challenges, and opportunities skin biopsy and beyond. Suppl Clin Neurophysiol. 2004; 57: 8–14.
- Martinez P, Aggio M, Rozenfeld P. High incidence of autoantibodies in Fabry disease patients. J Inherit Metab Dis. 2007; 30: 365–369.
- Germain DP, Charrow J, Desnick RJ, et al. Ten year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet. 2015; 52: 353-358.
- Linthorst GE, Hollak CE, Donker-Koopman WE, et al. Enzyme therapy for Fabry disease neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004; 66(4): 1589–95 (Epub 2004/10/02).
- Benichou B, Goyal S, Sung C, et al. A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol Genet Metab. 2009; 96(1): 4–12 (Epub 2008/11/22).
- Linthorst GE, Hollak CE, Donker-Koopman WE, et al. Enzyme therapy for Fabry disease neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004; 66(4): 1589–95.
- Aerts Johannes M, Groener Johanna E, Kuiper Sijmen, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. PNAS. 2008; 105(8): 2812–7.
- Schuller Y, Linthorst GE, Hollak CE, et al. Pain management strategies for neuropathic pain in Fabry disease--a systematic review. BMC Musc Disord. 2016; 16: 25.
- Hilz MJ, Brys M, Marthol H, et al. Enzyme replacement therapy improves function of C- Adelta- and Abeta-nerve fibers in Fabry neuropathy. Neurology. 2004; 62(7): 1066–72 (Epub 2004/04/14).
- Jardim LB, Gomes I, Netto CB, et al. Improvement of sympathetic skin responses under enzyme replacement therapy in Fabry disease. J Inherit Metab Dis. 2006; 29(5): 653–9 (Epub 2006/09/15).
- Mehta A, West ML, Pintos-Morell G, et al. Therapeutic goals in the treatment of Fabry disease. Genet Med. 2010; 12(11): 713–20.
- Politei JM, Bouhassira D, Germain DP, et al. Pain in Fabry Disease Practical Recommendations for Diagnosis and Treatment. CNS Neurosci Ther. 2016; 22(7): 568-576.
- Schiffmann R, Floeter MK, Dambrosia JM, et al. Enzyme replacement therapy improves peripheral nerve and sweat function in Fabry disease. Muscle Nerve. 2003; 28: 703–10.
- Hilz MJ, Brys M, Marthol H, et al. Enzyme replacement therapy improves function of C- Ad and Ab nerve fibers in Fabry neuropathy. Neurology. 2004; 62: 1066–72.
- Rombach SM, Smid BE, Bouwman MG, et al. Long term enzyme replacement therapy for Fabry disease effectiveness on kidney heart and brain. Orphanet J Rare Dis. 2013; 8: 47.
- Germain DP, Hughes DA, Nicholls K, et al. Treatment of Fabry's Disease with the Pharmacologic Chaperone Migalastat. N Engl J Med. 2016; 375(6): 545-555.