
Farber disease: A Fatal Childhood Disorder with Nervous System Involvement
Ichraf Kraoua1,2,3, Thouraya Ben Younes1,2,3*, Virginie Garcia4, Hanene Benrhouma1,2,3, Hedia Klaa1,2,3, Aida Rouissi1,2,3, Thierry Levade4,5, Ilhem Ben Youssef-Turki1,2,3
1Department of Child and Adolescent Neurology. National Institute Mongi Ben Hmida of Neurology. Tunis, Tunisia
2Research Laboratory LR18 SP04. National Institute Mongi Ben Hmida of Neurology. Tunis, Tunisia
3University of Tunis El Manar, Faculty of Medicine of Tunis. Tunis, Tunisia
4Cancer Research Center of Toulouse, INSERM UMR1037 and Université Paul Sabatier, Toulouse, France
5Laboratory of Metabolic Biochemistry, Federative Institute of Biology, CHU Purpan, Toulouse, France
Abstract
Farber Disease is an autosomal recessive inherited lysosomal storage disorder which is characterized by tissue accumulation of ceramide. It is caused by mutations within ASAH1 encoding for acid ceramidase. It represents a rare condition. Only twenty seven cases have been reported. Seven subtypes of Farber disease have been identified. The clinical presentation is characterized by the appearance of subcutaneous skin nodules, bone and joint deformities, and progressive hoarseness. Neurological symptoms as psychomotor delay or regression, hypotonia, seizures, and peripheral neuropathy were reported in some subtypes of Farber disease. The nervous system involvement is correlated to poor prognosis. In this study, we report on clinical, biochemical and molecular findings of two Tunisian siblings with Farber disease.
Introduction
Farber disease is a rare inborn lysosomal storage disorder characterized by accumulation of ceramide (N-acylsphingosine) in tissues, due to deficient activity of the acid ceramidase1. It is caused by mutations within ASAH12. It is associated with distinct clinical phenotypes3. Progressive neurologic involvement was reported in many cases2. To date, twenty seven cases were reported 4. Herein, we report on two additional cases of Farber disease belonging to a Tunisian family.
Case report
Patients are from consanguineous healthy parents from Tunisia during the period of 2015-2017. There was a family history of three maternal miscarriages, fulminant hepatopathy in a brother who died at the age of 10 years. The first patient was a 22-month-old boy. He was born after an uneventful pregnancy and delivery. Birth weight was 3.500 kg (50th - 85th percentiles). He had normal initial psychomotor development (head control at the age of 2 months, sitting at the age of 6 months). At the age of 6 months, he presented with hoarseness of the voice. Few months later, subcutaneous nodules appeared. Later on, we noticed decreased oral intake and no gain in motor skills. He was referred to our department at the age of 22 months. Examination at this age showed a normocephalic boy (50th -85th percentiles), a weight of 8.4 kg (below the 3rd percentiles), irritability, generalized hypotonia, brisk tendon reflexes, tongue fasciculations, nystagmus, hoarseness, and painful swelling of the interphalangeal and metacarpal joints. The fingers were held flexed at the intraphalangeal joints (figure 1). Fundus examination showed a bilateral cherry-red spot. Biological tests disclosed to hypertriglyceridaemia (2.31mmol / l; normal values <1.7mmol / l), high level of lactate dehydrogenase (1578 IU / L; normal values: 100-225 IU / L), and aspartate aminotransferase (51 IU / L; normal values<35 IU / L). Alanine aminotransferase and creatine phosphokinase level were normal (13 IU / L; normal values <45 IU / L). Brain MRI showed hypoplasia of the corpus callosum and diffuse cortical and subcortical atrophy. The results of nerve conduction studies and electromyography (EMG) were compatible with horn cell disease. The diagnosis of the classical form of Farber disease was suspected. The activity of acid ceramidase on peripheral leukocytes was found to be undetectable (0.05 nmol/h/mg; normal value: 9.8 nmol/h/mg). The gene sequencing of ASAH1 concluded to homozygosity for the c.107A>G pathogenic variation in exon 2 (p.Tyr36Cys) and presence of a homozygous polymorphic change in exon 10 (c.737T>C, p.Val246Ala). Our patient showed continuous worsening of his condition. He developed at 3 years of age seizures, respiratory and sleeping difficulties. He died at the age of 3 years 6 months.
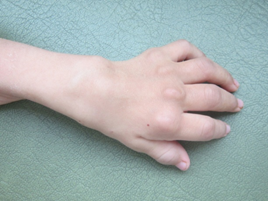
Figure 1: subcutaneous skin nodules near interphalangeal, metacarpal and wrist joints.
The second patient, the younger sister, who was already born at the time of the diagnosis of Farber disease in her brother, was 13 months old. She was born after an uneventful pregnancy by cesarean section secondary to bicicatricial uterus. A good Apgar score was recorded. Her birth weight was 3 kg (15th - 50th percentiles). She had a moderate psychomotor delay (head control at the age of 3 months, sitting at the age of 9 months, babbling at 12 months). At the age of 9 months, she presented with bilateral pain in the elbows extending to the interphalangeal and metacarpal joints. Examination showed a head circumference of 45 cm (15th -50th percentiles), a weight of 7.8 kg (3rd-15th percentiles), a length of 68 cm (below 3rd percentiles), irritability, hoarse weak cry, brisk tendon reflexes, bilateral Hoffman sign, knee contractures, and subcutaneous nodules near the metacarpal joints. Taking into account the family history, Farber disease was suspected. Biological tests (complete blood count, serum triglyceride and cholesterol, creatinine, aspartate aminotransferase, alanine aminotransferase and creatine phosphokinase), brain MRI, EMG, electrocardiogram, fundus examination, and abdominal ultrasound were normal. Ceramidase enzyme activity assay and genetic study were not performed. She was treated symptomatically with analgesics. The patient showed a continuous worsening of her condition over the following months. Aged 20, she developed a cerebellar syndrome and mystagmus. She died at the age of 2 years 6 months after respiratory infection.
Discussion
Our study illustrates additional cases of Farber disease with nervous system involvement and fatal issue.
The first case of Farber disease was described in 1957 by Sidney Farber5. It is a very rare inherited disease. Ninety six cases have been reported4. It is caused by deficiency of lysosomal acid ceramidase which is responsible for degradation of ceramide into sphingosine and free fatty acids within lysosomes6. Subsequently, there is an increased storage of ceramide in several organs and tissues3. This condition results from mutations in the ASAH1 gene and is inherited in an autosomal recessive manner.
The pathogenesis of Farber disease is largely unknown. Ceramide has been proposed to mediate apoptosis. Alterations of receptor-mediated apoptosis by ceramide accumulation in inflammatory cells may explain abnormal granuloma formation3. The pathogenic mechanisms of Farber Disease were studied using acid ceramidase mutant mouse model generated by deletion of the acid ceramidase signal peptide sequence. It was shown that deletion of the signal peptide sequence disrupts lysosomal targeting and enzyme activity, resulting in ceramide and sphingomyelin accumulation. Histiocytic infiltrations were observed in many tissues. The affected mice fail to thrive and die early7.
Seven subtypes of Farber disease have been identified according to age at onset, severity of the disease, and affected tissues. Type 1 represents the classical form of the disease. It includes patients with subcutaneous nodules, joint contractures, and voice hoarseness. Progressive neurologic involvement and lung disease were reported. Type 2 and 3 patients show only slight or no symptoms of central nervous system disease. Type 4 is associated with the “Neonatal-Visceral” variant. Neonates presented with severe organomegaly. Type 5 patients manifested by progressive neurological deterioration and seizures beginning at 1 to 2 1/2 years of life. Nodules and joint involvement are less severe in this type. Type 6 is termed “Combined Farber and Sandhoff Disease variant.” Type 7 patients have reduced glucocerebrosidase, galactocerebrosidase and ceramidase activities resulting from sphingolipid activator prosaposin deficiency6,8.
Our patients were diagnosed with type 1 Farber disease. First symptoms usually appear at 3-6 months of age. They include deformed joints, painful subcutaneous nodules, and progressive hoarseness due to laryngeal involvement6. The first symptom of Farber disease in our patients was noted during the first year of life, as reported in litterature. Involvement of the peripheral and central nervous system is observed in some subtypes of the disease. Psychomotor delay or regression, seizures, nystagmus, hypotonia, ataxia, loss of central white matter, or peripheral neuropathy have been reported2,8,9. Electromyogram shows sensory and motor demyelinating neuropathy or cell horn disease2. Both patients presented with central nervous system involvement represented essentially by psychomotor delay and regression. The older patient had also a peripheral nervous system involvement. Ophthalmological examination showed cherry red spot in some cases as seen in the first patient6,9.
Some biological anomalies like increased inflammatory marker, normocytic anemia, hyperferritinaemia, hypertriglyceridaemia, elevated aspartate aminotransferase levels, and high level of lactate dehydrogenase were reported10,11. The last three anomalies were noted in the first patient.
The diagnosis of Farber disease is confirmed by determination of acid ceramidase activity measured in cultured skin fibroblasts, white blood cells or amniocytes, by demonstration of granulomas with macrophages containing lipid cytoplasmic inclusions in subcutaneous nodules, or by determination of ceramide accumulation in tissues by chromatography or mass spectrometry3. The activity of acid ceramidase on peripheral leukocytes was found to be undetectable in our first patient.
A variety of mutations in the ASAH1 gene have been identified. About sixty different mutations have been found12. No clear phenotype-genotype correlation has been established8. Our patient was homoallelic for the p.Tyr36Cys substitution, which is reported to cause a rapid degradation of the mature enzyme13. This underlines the pathogenic role of this particular mutation in the development of a severe neurological phenotype.
The management of Farber disease is mainly palliative. It is focused on pain therapy, physical therapy and surgical correction of joint contractures. Bone marrow transplantation has shown some promising results on patients with minimal central nervous system involvement8. It may not be appropriate for patients with central nervous system involvement as ceramide neurotoxicity may not be reversible 3. Our patients were treated symptomatically with analgesics.
The early-onset neurovisceral form of Farber disease has a poor prognosis. As observed in the presently described family, patients die at approximately 2 years of age because of nervous system involvement or respiratory insufficiency, as observed in our cases3.
Conclusion
Farber disease is a rare and severe inherited disease. It could affect both central and peripheral nervous system which makes prognosis poor. It must be suspected in the presence of psychomotor retardation or psychomotor regression, particularly in the presence of subcutaneous nodules and hoarse voice. Early diagnosis helps prevent complications. Genetic diagnosis is necessary to confirm the phenotype and to start family screening. Multidisciplinary team involvement is crucial for the management of patients with Farber disease.
References
- Cvitanovic-Sojat L, Juraski R.G, Sabourdy F, Fensom A, Fumic K, Paschke E, et al. Farber lipogranulomatosis type 1–late presentation and early death in a Croatian boy with a novel homozygous ASAH1 mutation. Eur J Paediatr Neurol. 2011;15(2):171-173.
- Chedrawi AK, Al-Hassnan ZN, Al-Muhaizea M, Colak D, Al-Younes B, Albakheet A, et al. Novel V97G ASAH1 mutation found in Farber disease patients: unique appearance of the disease with an intermediate severity, and marked early involvement of central and peripheral nervous system. Brain Dev. 2012;34(5):400-404.
- Ehlert K, Frosch M, Fehse N, Zander A, Roth J, Vormoor J. Farber disease: clinical presentation, pathogenesis and a new approach to treatment. Pediatr Rheumatol. 2007;5:15.
- Zielonka M, Garbade SF, Kölker S, Hoffmann GF, Ries M. A cross-sectional quantitative analysis of the natural history of Farber disease: an ultra-orphan condition with rheumatologic and neurological cardinal disease features. Genet Med. 2018;20(5):524-530.
- Farber S, Cohen J, Uzman L. Lipogranulomatosis; a new lipo-glycoprotein storage disease. J Mt Sinai Hosp N Y. 1957;24(6):816-37.
- Al Jasmi F. A novel mutation in an atypical presentation of the rare infantile Farber disease. Brain Dev. 2012;34(6):533-535.
- Beckmann N, Kadow S, Schumacher F, Gothert JR, Kesper S, Draeger A, et al. Pathological Manifestations of Farber Disease in a New Mouse Model. Biol. Chem. 2018;399(10):1183-1202.
- Ehlert K, Levade T, Di Rocco M, Lanino E, Albert MH, Führer M, et al. Allogeneic hematopoietic cell transplantation in Farber disease. J Inherit Metab Dis 2019; 42(2):286-294.
- Cappellari AM, Torcoletti M, Triulzi F, Corona F. Nervous system involvement in Farber disease. J Inherit Metab Dis. 2016;39(1):149-150.
- Torcoletti M, Petaccia A, Pinto RM, Hladnik U, Locatelli F, Agostoni C, et al. Farber disease in infancy resembling juvenile idiopathic arthritis: identification of two new mutations and a good early response to allogeneic haematopoietic stem cell transplantation. Rheumatology (Oxford). 2014;53(8):1533-1534.
- Nivaggioni V, Cano A, Arnoux I, Michel G, Loosveld M. Early morphological diagnosis of Farber disease. Br J Haematol, 2016;175(2):189.
- Fabian P, Amintas S, Levade T, Medin JA. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J Rare Dis. 2018; 13(1):121.
- Bär J, Linke T, Ferlinz K, Neumann U, Schuchman EH, Sandhoff K. Molecular analysis of acid ceramidase deficiency in patients with Farber disease. Hum Mutat. 2001; 17(3):199-209.