
Multiple Exostosis Disease
Moustapha Niasse1*, Baïdy Sy Kane2, Kaba Condé3, Silly Touré4, Lamine Sarr5, Coumba Diouf1, Saïdou Diallo1
1Department of Rheumatology, University Hospital Aristide Le Dantec of Dakar, Senegal
2Department of Internal Medicine, University Hospital Aristide Le Dantec of Dakar, Senegal
3Department of Rheumatology, University Hospital Ignace Deen of Conakry, Guinea
4Department of Stomatology and Maxillofacial surgery, University Hospital Aristide Le Dantec of Dakar, Senegal
5Department of Orthopaedics and Traumatology, University Hospital Aristide Le Dantec of Dakar, Senegal
Abstract
Multiple exostosis disease is one of the hereditary diseases with autosomal dominant transmission. It is characterized by the proliferation of bone protuberances, especially located in the metaphysis of long bones.
Since 1993, advances have been noted in knowledge of the pathophysiology of this disease, in particular with the discovery of the mutation of EXT genes, found in 80% of multiple exostosis disease. These genes, tumor suppressors, code for proteins involved in the synthesis of heparan sulfates. The deficiency in quantity and quality of heparan sulfates leads to changes in certain metabolic processes, which leads to the development of ectopic growth plaques. This is responsible for the development of exostosis, but also for the low longitudinal growth of long bones. The disease phenotype may also associate abnormalities in the shape and length of long bones, such as the typical “Bessel Hagen” deformity. Clinically, bone masses are often painless. The rare complication (2 to 5% of cases) but the most feared is the transformation into chondrosarcoma, which motivates regular clinical and radiological monitoring of these patients. Treatment, mainly surgical, is indicated in case of symptoms (pain, increased exostosis volume after the end of growth, compression of neighboring organs.
Introduction
Multiple exostosis disease, first described by French surgeon Alexis Boyer in 1814, is a rare genetic disease with autosomal dominant transmission1. There is a family history in about 60% of cases2. Exostosis is composed of bone tissue with a peripheral cortex, central spongy bone, and a cartilaginous cap2. All bones developing by endochondral ossification can be affected (long bones, vertebrae, medial portion of the clavicle and bone of the base of the skull)3. Bones that develop by membrane ossification are not affected (flat bones, lateral portion of the clavicle), except in special cases3. Exostosis develops during childhood. They stabilize with progressive regression of the cartilage cap after the end of growth4.
Epidemiology
Multiple exostosis disease is a rare condition with an estimated prevalence of 1/500,000 in the western population5, 6. It is less reported in North Africa7, 8. In sub-Saharan Africa, only 20 observations to our knowledge were described between 1999 and 2018 (table below)9, 10, 11, 12, 12, 13, 14, 15.
Table 1. Summary table of cases of multiple exostosis disease reported in Sub-Saharan Africa
| AUTHORS | YEAR | COUNTRY | NUMBER OF CASES |
|---|---|---|---|
| Govender S [9] | 1999 | South Africa | 2 |
| Bamba I 10] | 2002 | Ivory Coast | 1 |
| Ntsiba H [11] | 2002 | Congo | 4 |
| Dao F [12] | 2013 | Burkina | 1 |
| Diallo S [13] | 2016 | 1 | |
| Niasse M [14] | 2018 | Senegal | 10 |
| Bukara E [15] | 2018 | Rwanda | 1 |
Physiopathology
Genetic
Multiple exostosis disease is associated with a mutation of the genes EXT1 (chromosome 8q23-q24)16 and/or EXT2 (chromosome 11p11-p13)17, found in 80% to 90% of cases. An EXT3 gene (chromosome 19) has also been described, but its responsibility for the development of the disease remains to be determined18. EXT genes are tumor suppressors: the loss of heterozygosity related to their mutation would be an inductive event in the development of exostosis19. These genes code for transmembrane type II glycoproteins, exostosin 1 and 220, 21, which have glycosyltransferase action2, 21. These two proteins, mainly located in the endoplasmic reticulum form a Golgi-localized heterooligomeric complex that catalyzes the polymerization of heparan sulfate (HS).
The consensus would be that exostosin 1 would have the most important synthesis activity and exostosin 2 would stabilize the heterodimer in the Golgi apparatus. The mutation of the EXT1 and EXT2 genes is therefore responsible for the decrease in the concentration of heparan sulfates. Moreover, the exostosis biopsy study of patients with multiple exostosis disease showed a higher concentration in heparanase cartilage compared to healthy controls and solitary exostosis2. This also suggests an increase in the degradation of heparan sulfates22, 23.
Consequence of heparan sulfate deficiency
During the chondrocyte maturation phase, chondrocytes go through the prehypertrophic, hypertrophic and then enter apoptosis2. They are organized in regular columns in the metaphysis allowing longitudinal growth of long bones2, 24.
Heparan sulfate chains have a co-receptor role in many metabolic pathways, such as Fibroblast Growth Factor (FGF), Tumor Growth Factor (TGF), Bone Morphogenetic Protein (BMP), Wnt, Indian Hedgehog (Ihhh), PTHrp25. The integrity of these signaling pathways is essential for the different phases of chondrocyte maturation at the growth plate during endochondral ossification24. The pathophysiological hypothesis is that the decrease in HS on the surface of chondrocytes would slow down their differentiation into hypertrophic chondrocytes, giving them a proliferation advantage at the same time. This proliferation would lead to a lack of ossification of the perichondrium, explaining the growth of ectopic cartilage26.
The distribution of Ihh in the growth plate is dependent on the concentration of heparan sulfate: the mutation of EXT1 induces an increase in the diffusion zone of Ihh with extension towards the perichondrium. Moreover, Ihh causes an increase in chondrocyte proliferation and a decrease in differentiation into hypertrophic chondrocytes27. This may be related to the fact that heparan sulfate can bind to Hedgehog ligands and modulate their diffusion in the extracellular medium: the decrease of heparan sulfate concentration would lead to a decrease in the possibility of ligand binding and activation of the Hedgehog signal in patients with multiple exostosis disease28.
Heparan sulfates would limit the diffusion zone of Wnt (a major actor in bone and cartilage formation) by facilitating the binding of Wnt to its receptor28. Thus, the action of the Wnt signal is reduced compared to healthy individuals, which could partly explain the abnormalities in the organization of the growth plate in affected patients28, 29.
For the FGF signal, the decrease in heparan sulfate concentration results in a marked decrease in the activation of the MAP kinase pathway, which are intracellular mediators essential to the metabolic pathway of FGF29.
The analysis of BMP receptors and BMP expression in the cartilage layer of exostoses is similar to the rates found in the growth plate. This could explain the presence of a prolonged proliferation of exostosis in affected patients30.
The presence of heparanase has been identified in exostosis chondrocytes of patients with multiple exostosis disease2.
Finally, the position in the growth plate of mutated chondrocytes could be important in the development of exostosis. In a zebrafish model, the implantation of mutated chondrocytes dak-/- (equivalent to EXT) in the center of the growth plate is without consequence: the mutated cells are placed in the regular columns, "reoriented" by the adjacent normal cells. Inversely, by implanting these same mutated cells on the periphery of the growth plate, dak-/- chondrocytes grow perpendicular to the growth plate31. This observation highlights the role of the cartilage-perichondrium transition zone in the development of bone exostosis and may partly explain the absence of development of intracartilage lesions, such as enchondromes in presence of EXT mutations32.
Diagnosis
Positive diagnosis
The diagnosis of multiple exostosis disease is often early. It is performed in the majority of cases before the age of 5 years, because of the accessible nature of the bone masses, which are however painless5, 8. The most frequent locations are mainly active metaphyses of bones developing by endochondral ossification: mainly in the distal femur, fibula and proximal tibia, proximal humerus (Figure 1).
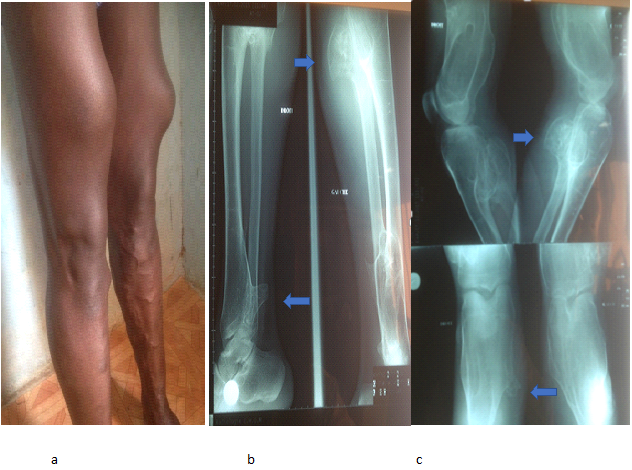
Figure 1: Multiple exostosis diseases: clinical (a) and radiological images (b, c) of exostosis in the metaphyses of the bones of the lower limbs
Some locations are rare but potentially serious, such as ribs, rachis or pelvis, due to proximity to important structures such as the spinal cord, lungs, heart or urogenital organs14, 33(Figure 2).
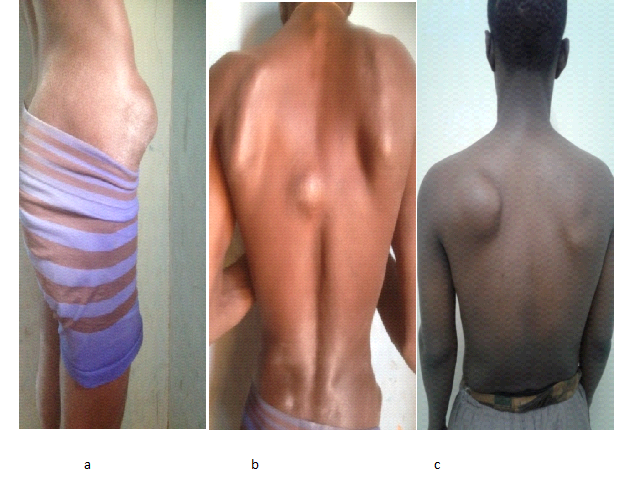
Figure 2: Multiple exostosis disease : exostoses localized to the axial skeleton
Patients are often small, with short limbs, deformed in two-thirds of patients1. The most common orthopedic complications include bone synostosis, limb inequality, varus or valgus deformations in the knees. Limb inequalities are the result of bone shortening. Dysharmony in the length of the forearm bones may be responsible for arcature of one or both forearm bones, an ulnar bote hand with ulnar translation of the carpus or subluxation/luxation of the radial head2, 33, 34. The typical deformity mainly affecting ulna is called Bessel Hagen disease2, 33(Figure 3).
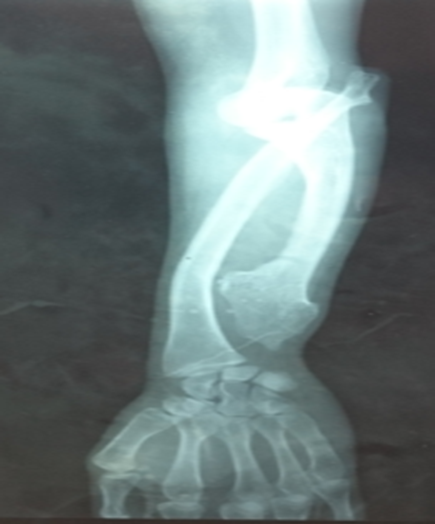
Figure 3: Multiple exostosis disease : Bessel Hagen disease (arched deformation of the ulna by shortening the ulna). Dislocation of the upper joint between the ulna and radius.
The diagnosis of exostosis is essentially radiological34. In the typical form, histology is not required34. The radiological forms of exostosis are multiple but can be individualized into 3 types: pedicle forms (narrow implantation and always inclined towards the diaphysis), sessile forms, characterized by a wide implantation on the metaphysis and diaphysis and cauliflower forms34.
The tomodensitometry shows continuity between the spongy metaphyseal bone spans and the center of the exostosis (Figure 4). This element is pathognomonic, however, it is often missing on standard radiographs34.
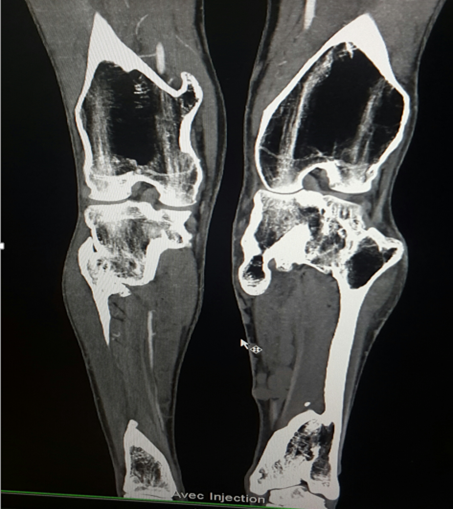
Figure 4: Multiple exostosis disease: continuity between the spongy metaphyseal bone spans and the centre of exostosis.
Magnetic resonance imaging is the best radiological technique to assess the relationship of exostosis with neighboring anatomical structures and to evaluate the cartilage cap. The mineralized parts are hyposignal in all sequences, while the non-mineralized parts with a high water content are hyposignal T1 and hypersignal intense T2. These characteristics allow the precise measurement of the thickness of the cartilage cap by determining its limit with the overlying muscles34.
Differential diagnosis
Multiple exostosis disease can be confusing due to certain diseases, in particular:
-Metachondromatosis, which is an autosomal dominant disease induced by the mutation of the PTPN11 gene. It is characterized by exostoses that move towards the joint35. The disease progresses to spontaneous regression35.
-Langer-Giedon syndrome, which is linked to a mutation of the EXT1 and TRPS1 genes. Clinical presentation combines multiple exostoses, mental retardation, abnormalities of the skull and face and digital2.
-Potocki-Shaffer syndrome, which is linked to a mutation of the EXT2 and ALX4 genes. Clinical presentation combines multiple exostoses, skull ossification deficiency (enlarged parietal foramen), skull-facial dysostosis and mental retardation36.
-Progressive ossifying fibrodysplasia, which is linked to a mutation in the ACVR1 gene (activin receptor) inducing activation of the ALK2 receptor at BMP1. This disease combines an overall ossification deficiency, a short femoral neck, an enlargement of the metaphyses and the presence of exostosis, especially of the proximal tibia37.
Prognosis
Depending on its location and volume, exostosis can be responsible for complications that can be severe: osteo-articular complications (osteo-articular deformities, bone fractures, osteomyelitis), compressions of adjacent structures (nerve compressions, vascular compressions, intra-thoracic compressions)38, 39, 40, 41, 42, 43, 44.
The most serious complication is malignant transformation, which can occur in 2 to 5% of cases37. It is a chondrosarcoma in 90% of cases37. The predictive signs of malignant degeneration are the increase in size of exostoses, especially after the end of growth, the presence of local inflammatory signs, an increase in the cartilaginous cap thickness (greater than 2 cm in adults or 3 cm in children) (figure 5)45. Locations with pelvic and scapular belts are more susceptible to degeneration46.
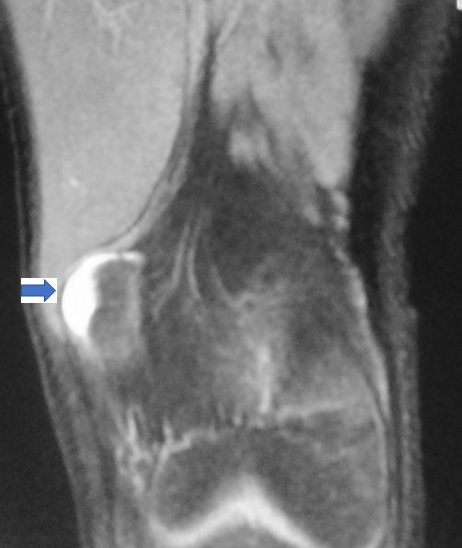
Figure 5: Magnetic resonance imaging in multiple exostosis disease: Increase of the thickness of the cartilage cap [34]
Processing and monitoring
The treatment proposed for multiple exostosis disease is mainly surgical. Surgery should be offered in case of symptoms (pain, increases in exostosis volumes after growth has ceased, compressions of neighboring organs)34, 47.
In case of osteochondromas stability, regular close monitoring is necessary due to the risk of malignant degeneration34. Computed tomography and magnetic resonance tomography (MRI) are an important part of this monitoring34.
Conclusion
Multiple exostosis disease is a rare disease, more reported in Western literature. It results in exostosis in the metaphyses of long bones and more rarely in the axial skeleton. The most feared complication is the malignant transformation into chondrosarcoma, which requires close monitoring of these patients. Despite the progress of knowledge about the disease, current treatments remain purely symptomatic. However, there are many research pathways, and the target of growth plate actors (Ihh, BMP, FGF) and heparan sulfate metabolism could generate therapeutic leads.
References
-
- Solomon L. Hereditary multiple exostosis. Am J Hum Genet. 1964; 16: 351-363.
- Robin F, Ropars M, Violas P, et al. Multiples Hereditary multiple. Rev Rhum monographies. 2018; 86(2): 84-91.
- Ducy P. Genetic control of skeletogenesis. Médecine/Sciences. 2001; 17: 1242-1251.
- Legeai-Mallet L, Munnich A, Maroteaux P, et al. Incomplete penetrance and expressivity skewing in hereditary multiple exostoses. Clin Genet. 1997; 52: 12-16.
- Rambeloarisoa J, El Guedja M, Legeai-Malletd L, et al. Hereditary multiple exostoses with 40 years follow-up: case report. Rev Med Interne. 2002; 23(7): 657-664.
- Sonne-Holm E, Wong C, Sonne-Holm S. Multiple cartilaginous exostoses and development of chondrosarcomas-a systematic review. Dan Med J. 2014; 61(9): A4895.
- Sfar S, Abid A, Mahfoudh W, et al. Genetic analysis of hereditary multiple exostoses in Tunisian families: a novel frame-shift mutation in the EXT1 gene. Molecular Biology Reports. 2009; 36(4): 661-667.
- Ameziane L, EL Bardouni A, Mahfoud M, et al. Exostosis of limbs. Médecine du Maghreb. 1999; 75: 33-35.
- Govender S, Parbhoo AH. Osteochondroma with compression of the spinal cord. A report of two cases. J Bone Joint Surg Br. 1999; 81(4): 667-669.
- Bamba I, Sie-Essoh JB, Aka Kacou D, et al. Multiple exostosis. Review of the literature. Apropos of a case disclosed during an injury. Bull Soc Pathol Exot. 2002; 95: 83-85.
- Ntsiba H, Bazébissa R. Multiple exostoses. The first 4 Congolese cases. Bull Soc Pathol Exot. 2002; 95(1): 20-22.
- Dao F, Yonaba C, Diallo O, et al. Bessef Hogen’S disease or illness exostosis’s disease with spinal cord compression: about a case in the service of the paediatric CHU Ouedraogo, Ouagadougou. Mali Médical. 2013; 1: 47-48.
- Diallo S, Niasse M, Diallo R, et al. Hereditary multiple exostoses: A case-report. Joint Bone Spine. 2016; 83 :455.
- Niasse M, Kane BS, Garba MS, et al. Multiple exostosis disease: study of two senegalese multiplex families. Rev Mar Rhum. 2018; 44: 46-53.
- Bukara E, Buteera AM, Karakire R, et al. Osteoblastic Osteosarcoma Arising beneath an Osteochondroma in an 11-Year-Old Male with Multiple Hereditary Exostoses. Case Rep Orthop. 2018; 12: 1-8.
- Cook A, Raskind WH, Blanton SH, et al. Genetic Heterogeneity in Families with Hereditary Multiple Exostoses. Am J Hum Genet. 1993; 53: 71-79.
- Wuyts W, Van Hul W. Molecular basis of multiple exostoses: mutations in theEXT1 and EXT2 genes. Hum Mutat. 2000; 15: 220-227.
- Le Merrer M, Legeai-Mallet L, Jeannin PM, et al. A gene for hereditary multipleexostoses maps to chromosome 19p. Hum Mol Genet. 1994; 3: 717-722.
- Porter DE, Simpson A. The neoplastic pathogenesis of solitary and multipleosteochondromas. J Pathol. 1999; 188: 119-125.
- Cheung PK, McCormick C, Crawford BE, et al. Etiological point muta-tions in the hereditary multiple exostoses gene EXT1: a functional analysisof heparan sulfate polymerase activity. Am J Hum Genet. 2001; 69: 55-66.
- Legeai-Mallet L, Bonaventure J. Exostosins: proteins involved in the biosynthesis of heparan sulfates. Medecine/sciences. 2002; 18: 23-25.
- McCormick C, Duncan G, Goutsos KT, et al. The putative tumor suppres-sors EXT1 and EXT2 form a stable complex that accumulates in the Golgiapparatus and catalyzes the synthesis of heparan sulfate. Proc Natl Acad Sci. 2000; 97: 668-673.
- Trebicz-Geffen M, Robinson D, Evron Z, et al. The molecular and cel-lular basis of exostosis formation in hereditary multiple exostoses: hereditary multiple exostoses. Int J Exp Pathol. 2008; 89: 321-331.
- Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003; 423: 332.
- Häcker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005; 6: 530-541.
- Jones KB, Piombo V, Searby C, et al. A mouse model of osteochondromage-nesis from clonal inactivation of Ext1 in chondrocytes. Proc Natl Acad Sci. 2010; 107: 2054-2059.
- Koziel L, Kunath M, Kelly OG, et al. Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Dev Cell. 2004; 6: 801-813.
- Alman BA. Multiple hereditary exostosis and hedgehog signaling: implicationsfor novel therapies. JBJS. 2009; 91: 63-67.
- Lin X, Perrimon N. Role of heparan sulfate proteoglycans in cell-cellsignaling in Drosophila. Matrix Biol J Int Soc Matrix Biol. 2000; 19: 303-307.
- Cuellar A, Inui A, James MA, et al. Immunohistochemical localizationof bone morphogenetic proteins (BMPs) and their receptors in solitaryand multiple human osteochondromas. J Histochem Cytochem. 2014; 62: 488-498.
- Clément A, Wiweger M, von der Hardt S, et al. Regulation of zebrafish skele-togenesis by ext2/dackel and papst1/pinscher. PLoS Genet. 2008; 4: e1000136.
- Matsumoto K, Irie F, Mackem S, et al. A mouse model of chondrocyte-specific somatic mutation reveals a role for Ext1 loss of heterozygosityin multiple hereditary exostoses. Proc Natl Acad Sci. 2010; 107: 10932-10937.
- Jäger M, Westhoff B, Portier S, et al. Clinical outcome and genotype inpatients with hereditary multiple exostoses. J Orthop Res. 2007; 25: 1541-1551.
- Moatassim Billaha N, Idrissia MM, Idrissi Kaitounib R, et al. Imaging of solitary exostoses. Feuillets de radiologie. 2013; 53: 11-20.
- Fisher TJ, Williams N, Morris L, et al. Metachondromatosis: more than just multiple osteochondromas. Child Orthop. 2013; 7: 455-464.
- Pannier S, Legeai-Mallet L. Hereditary multiple exostoses andenchondromatosis. Best Pract Res Clin Rheumatol. 2008; 22: 45-54.
- Deirmengian GK, Hebela NM, O’Connell M, et al. Proximal tibial osteochon-dromas in patients with fibrodysplasia ossificans progressiva. J Bone Jt Surg. 2008; 90: 366-374.
- Kalyani R, Prabhakar K, Gopinath B, et al. Multiple Heriditary Exostoses in a Family for Three Generation of Indian Origin with Review of Literature. Journal of Clinical and Diagnostic Research. 2014; 8(10): LD01-LD03.
- Nasr B, Albert B, David CH, et al. Exostoses and vascular complications in the lower limbs: two cases report and review of literature. Ann Vasc Surg. 2015; 29 (6): 1315-1319.
- Stieber JR, Dormans JP. Manifestations of hereditary multiple exostoses. J Am Acad Orthop Surg. 2005; 13: 110-120.
- Toumia S, Ghnayaa H, Essid A, et al. Hereditary multiple exostosis revealed by deep vein and arterial popliteal thrombosis. Rev Med interne. 2010; 31: e7-e10.
- Zaijun L, Xinhai Y, Zhipeng W, et al. Outcome and prognosis of myelopathy and radiculopathy from osteochondroma in the mobile spine: a report on 14 patients. J Spinal Disord Tech. 2013; 26(4): 194-199.
- Kane BS, Niasse M, Akpo G, et al. Disseminated masses. Rev Med interne. 2017; 38: 562-563.
- Niasse M, Kane BS, Mbodji MM, et al. Painless outgrowth bones: Multiple exostosis diseases. Presse Med. 2016; 45: 710-12.
- Cohen MM. The new bone biology : pathologic, molecular and clinical correlated. Am J Med Genet A. 2006; 140: 2646-2706.
- Mota López A, García-Roméu García C, Sanjose Pacheco F, et al. Review of exostosing disease apropos of the study of a family with 14 carriers of multiple exostoses. Rev Clin Esp. 1977; 15: 144: 350-360.
- Czajka CM, DiCaprio MR. What is the Proportion of Patients With Multiple Hereditary. Clin Orthop Relat Res. 2015; 473: 2355-22361.