
Takayasu's arteritis - a comprehensive review
H.S. Natraj Setty*, J.R. Vijaykumar, C.M Nagesh, Shivanand S Patil, Santhosh Jadav, T.R. Raghu, C.N. Manjunath
Sri Jayadeva Institute of Cardiovascular Sciences and Research, Bengaluru, Karnataka, India
Abstract
Takayasu’s arteritis (TA) is a chronic inflammatory disease of unknown aetiology. The mechanism of this disease is not exactly defined. The inflammatory process is generally (but not exclusively) initiated in the second or third decade of life through the actions of non-specific inflammatory cells. As the disease progresses, fibrotic stenosis occurs in aorta and its main branches. The consequence of this inflammatory process can be stenosis, thrombosis, dilatation or aneurysm formation in aorta and/ or its branches. Majority of cases have been observed in Asia, Africa, and Latin America. In Asia, its incidence (2.69 in a million per year) has been reported to be 100 times higher than in Europe and North America. Because of the delay in diagnosing the disease, patients often experience claudication, absence of pulses, hypertension, myocardial infarction (MI), and cerebrovascular accidents (CVAs). Accurate and early diagnosis of TA can reduce the economic, social, and psychological burdens. Considering the fact that classical TA has mainly been described in Asia.
Background
Takayasu arteritis (TA) is a large vessel vasculitis (LVV) characterized by granulomatous inflammation of the vessel wall with an unknown etiopathogenesis. TA predominantly affects young females during the second or third decades of life and mainly involves the aortic arch and its primary branches, ascending aorta, thoracic descending aorta and abdominal aorta (Figure A and B). Early in the disease course, inflammation of the involved arteries progresses, resulting in segmental stenosis, occlusion, dilatation and/or aneurysm. This may cause extremity pain, claudication, bruits, absent or diminished pulses and loss of blood pressure. TA generally follows an insidious course, however, presentation with acute visual loss or stroke may also occur1,2. TA may show different patterns of arterial involvement, disease expression and prognosis in different regions of the world3,4. Multiple genetic factors were recently shown by a whole-genome approach in TA and an association between the extent of vascular involvement and the major genetic risk factor HLA-B*52 was reported in Turkish TA patients, suggesting that genetic factors might influence disease severity5,6. Yamamoto described the case of a 45 year old man with persistent fever who developed impalpable upper limb and carotid pulses associated with weight loss and dyspnoea. Takayasu, professor of ophthalmology at Kanazawa University Japan, presented the case of a 21-year-old woman with characteristic fundal arteriovenous anastamoses. Onishi and Kagosha each described similar cases associated with absent radial pulses7. The first postmortem case of a 25-year-old woman demonstrated panarteritis and suggested that the fundal appearances resulted from retinal ischaemia8. Shimizu and Sano summarised the clinical features of this “pulseless disease”9. Incidence of Takayasu arteritis is rare, but most commonly seen in Japan, South East Asia, India, and Mexico. In 1990, it was included in the list of intractable diseases maintained by the Japanese government, and to date 5000 patients have been registered (Table 1). A study of North American patients by Hall et al found the incidence to be 2.6/million/year10. The UK incidence is unknown.
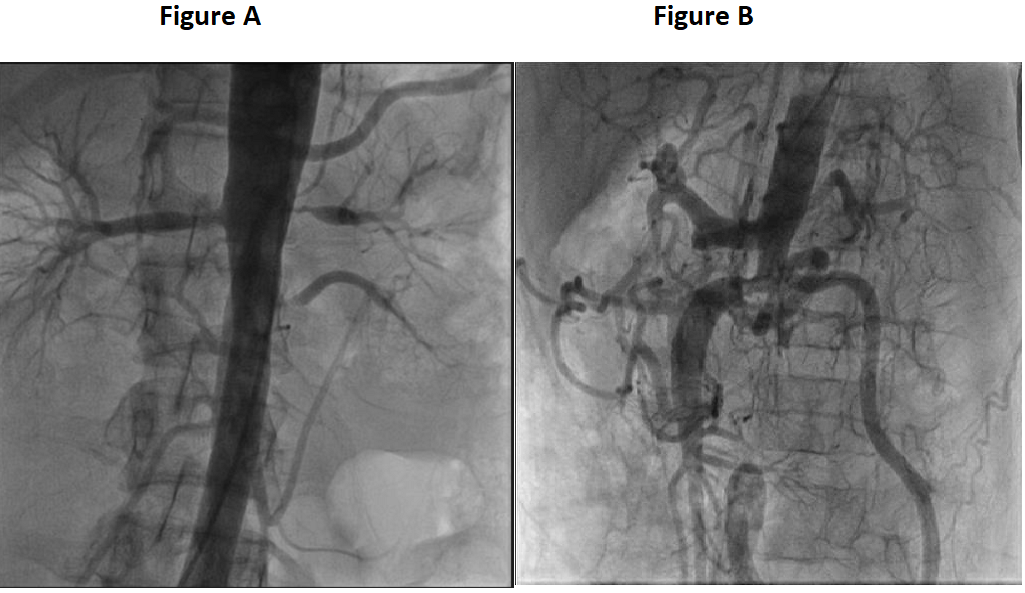
Images of takayasu’s arteritis with various clinical manifestations
Figure A: Shows left renal artery and accessory renal artery stenosis
Figure B: Shows abdominal aortic aneurysm
Table 1:1990 ACR criteria for the classification of Takayasu arteritis37
| Criteria | Definition |
|---|---|
| Age at disease onset <40 years | Development of symptoms or findings related to Takayasu arteritis at age <40 years |
| Claudication of extremities |
Development and worsening of fatigue and discomfort in muscles of 1 or more extremity while in use, especially the upper extremities |
| Decreased brachial artery pulse | Decreased pulsation of 1 or both brachial arteries |
| Blood pressure difference >10 mm Hg | Difference of >10 mm Hg in systolic blood pressure between arms |
| Bruit over subclavian arteries or aorta | Bruit audible on auscultation over 1 or both subclavian arteries or abdominal aorta |
| Arteriogram abnormality |
Bruit audible on auscultation over 1 or both subclavian arteries or abdominal aortaArteriographic narrowing or occlusion of the entire aorta, its primary branches, or large arteries in the proximal upper or lower extremities, not caused by arteriosclerosis, fibromuscular dysplasia, or similar causes; changes usually focal or segmental |
A diagnosis of Takayasu arteritis requires that at least 3 of the 6 criteria are met.
Pathophysiology
Takayasu arteritis is an inflammatory disease of largeand mediumsized arteries, with a predilection for the aorta and its branches. Advanced lesions demonstrate a panarteritis with intimal proliferation. Lesions produced by the inflammatory process can be stenotic, occlusive, or aneurysmal. All aneurysmal lesions may have areas of arterial narrowing. Vascular changes lead to the main complications, including hypertension, most often due to renal artery stenosis (Figure C and D) or, more rarely, stenosis of the suprarenal aorta; aortic insufficiency due to aortic valve involvement; pulmonary hypertension; and aortic or arterial aneurysm. Congestive heart failure is a common finding, much more so than dilated cardiomyopathy, myocarditis, and pericarditis, which also have been reported. Macroscopically, in the chronic phase, the aorta is thickened secondary to fibrosis of all three vessel layers. The lumen is narrowed in a patchy distribution, often affecting multiple areas. If disease progression is rapid, fibrosis can be inadequate with subsequent aneurysm formation. The intima may be ridged, with a “tree bark” appearance, a feature common to many aortitides11. Microscopically, the vasculitis may be divided into an acute florid inflammatory phase and a healed fibrotic phase. In the acute phase a vasa vasoritis is seen in the adventitia. The media is infiltrated by lymphocytes and occasional giant cells with neovascularisation. Mucopolysaccharides, smooth muscle cells, and fibroblasts thicken the intima. In the chronic phase there is fibrosis with destruction of elastic tissue. Similar histopathological findings are also seen in giant cell arteritis; therefore, biopsy results may not differentiate between these two vasculitides. Clinical manifestations usually allow correct diagnosis12.
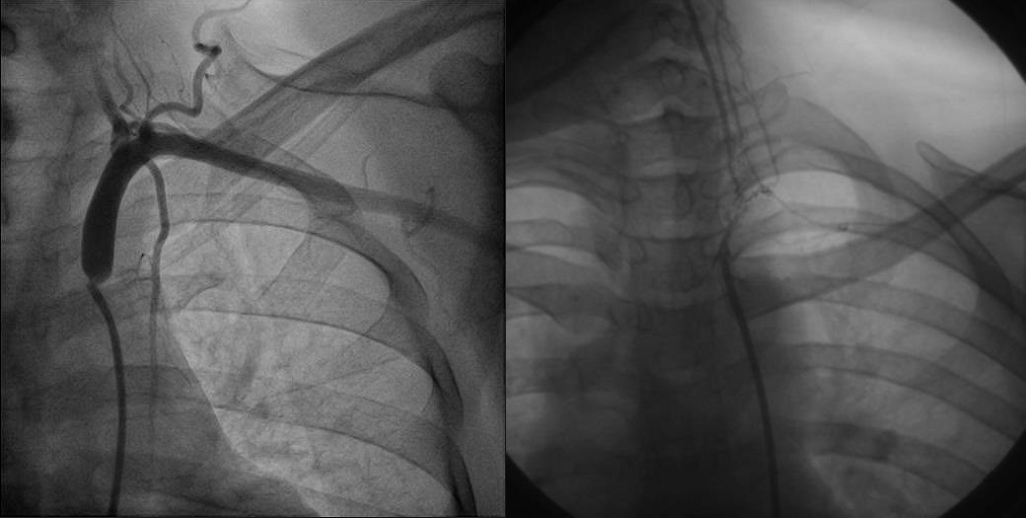
Figure C: Shows left Subclavian artery stenosis
Figure D: shows carotid artery stenosis.
Etiology
The etiology of TA is still not known13. Similarly, exact pathogenic sequence of the disease is yet to be established. However, a hypothesis has been developed where 65kDa heat-shock protein in the aortic tissue gets stimulated by unknown stimulus leading to the induction of major histocompatibility class I chain-related A (MICA) located on the vascular cells14. TA is a chronic granuloumatous panarteritis of large sized arteries, classically involving the aortic arch, but one third of the cases also affect the remainder of the aorta and its branches, and pulmonary arteries15. The gross morphologic examination, in most of the cases, irregular thickening of the aortic and its branch vessel wall with intimal wrinkling is seen16. When aortic arch is involved, the orifices of aortic branch vessels to the upper portion of the body may be markedly narrowed or even obliterated. The coronary and renal arteries may be equally affected (Figure E). Histological findings may range from an adventitial mononuclear infiltrate with perivascular cuffing of the vasa vasorum (channels supplying blood vessels) to marked mononuclear inflammation the media17. Aortic root involvement may lead to dilatation and aortic valve insufficiency18. Myocardial infarction may result in narrowing of coronary ostia19.
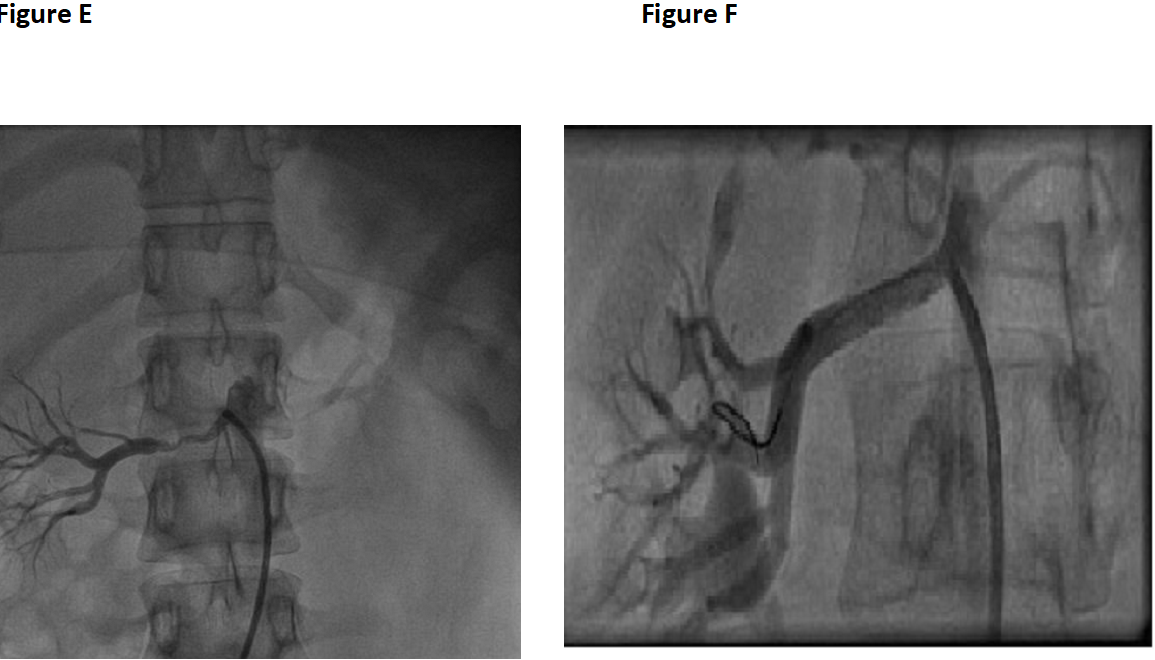
Figure E: Shows right renal artery stenosis
Figure F: Shows PTA to right renal artery
Clinical features
⇒ Diminished or absent pulses in 84–96% of patients associated with limb claudication and blood pressure discrepancies.
⇒ Vascular bruits in 80–94% of patients, often multiple, and particularly affecting the carotids, subclavian, and abdominal vessels20.
⇒ Hypertension in 33–83% of patients21, generally reflecting renal artery stenosis, which is seen in 28–75% of patients22.
⇒ Takayasu retinopathy in up to 37% of patients23.
⇒ Congestive cardiac failure associated with hypertension, aortic regurgitation, and dilated cardiomyopathy24.
⇒ Neurological features secondary to hypertension and/or ischaemia, including postural dizziness, seizures, and amaurosis.
⇒ Pulmonary artery involvement in 14–100% of patients, depending on the method used to assess pulmonary vasculature. Oligaemic lung fields on plain chest x ray correlate with pulmonary vasculopathy in approximately a third of cases. Pulmonary artery disease shows little correlation with the systemic pattern of arterial involvement25, but can be useful in the differential diagnosis by helping to confirm Takayasu arteritis.
Serological markers
No reliable serological marker of disease course has been identified. In the past, ESR normalisation was used as a remission criterion, but recently it has been shown that it has a low specificity and sensitivity, because ESR is raised in nearly half of the patients in clinical remission, while it is normal in 28% of the patients with active disease26. Serum concentrations of interleukin-6 and regulated on activation normal T cell expressed and secreted (RANTES) are elevated in patients with active disease and concentrations parallel to the activity of the disease27. Similarly, matrix metalloproteinase-3 and metalloproteinase-9 levels can be used as activity markers, whereas high serum concentrations of metalloproteinase-2 can suggest the presence of Takayasu’s arteritis without any relation with the activity of the disease28.
Radiological findings
The American College of Rheumatology included arteriogram abnormalities in the diagnostic criteria of the disease. Angiography is the gold standard for evaluation of vascular lesions; in particular panangiography allows a correct assessment of the extension of the disease, which correlates with its severity. Some authors underline the high incidence of coronary involvement in Takayasu’s arteritis (15%) and recommend to perform a coronarographic exam29. Assessment of pulmonary vasculature by angiography is not universally recommended being reserved for patients with symptoms of pulmonary hypertension30. However, non-invasive techniques (i.e. lung scan) have demonstrated lung perfusion abnormalities in about two thirds of asymptomatic patients31. Angiography allows a topographic classification which correlates anatomic involvement, clinical manifestations and prognosis. Angiography, however, is an invasive method, it is not able to differentiate active from burned-out lesions and exposes the patients to risks connected with radiation and the contrast medium. In the early phase of Takayasu’s arteritis, the thickening of vascular wall of the aorta or pulmonary artery can be detected by computed tomography (CT) or nuclear magnetic resonance (NMR). CT, combined with injection of contrast medium (the so-called angio-CT), allows to study wall inflammation but it has a low spatial resolution and cannot evaluate medium-size arteries32. NMR is a good alternative method to CT, especially angio-NMR, which is equally or more sensitive than angiography in detecting lesions of large vessels. However, it is less sensitive in detecting smaller branch involvement and may overestimate the degree of stenosis in renal and subclavian arteries33. More recently, fluorodeoxyglucose positron emission tomography (FDG-PET) has been proposed as a promising technique in the early diagnosis and assessment of response to treatment in Takayasu arteritis as well as in other large vessel vasculitis, because it seems to be a more sensitive method than conventional clinical methods in measuring continuing vascular inflammation34. Finally, color Doppler ultrasonography plays an important role for screening, detection and follow-up of carotid (Figure F and G) and subclavian arteries where it is easy to discriminate between atherosclerotic and inflammatory lesions35.
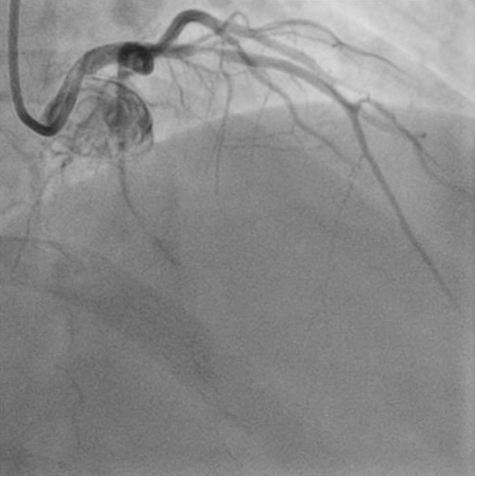
Figure G: Shows left anterior descending(LAD)coronary artery stenosis
Classification
Classification of Takayasu’s arteritis has been made to classify the disease on the basis of angiographic findings (Table 2). Ishikawa clinical classification and diagnostic criteria of Takayasu arteritis shown in Table 3 and Table 4. The early system, revised by Lupi-Herrera et al in 197736., has been superseded by the new classification of Takayasu arteritis. These systems are useful in that they allow a comparison of patient characteristics according to the vessels involved and are helpful in planning surgery.
Table 2:New angiographic classification of Takayasu arteritis, Takayasu conference 199438
| Type | Vessel involvement |
|---|---|
| Type I | Branches from the aortic arch |
| Type IIa | Ascending aorta, aortic arch and its branches |
| Type IIb | Ascending aorta, aortic arch and its branches, thoracic descending aorta |
| Type III | Thoracic descending aorta, abdominal aorta, and/or renal arteries |
| Type IV | Abdominal aorta and/or renal arteries |
| Type V | Combined features of types IIb and IV |
According to this classification system, involvement of the coronary or pulmonary arteries should be designated as C (+) or P (+), respectively.
Table 3:Ishikawa clinical classification of Takayasu arteritis39
| Group | Clinical features |
|---|---|
| Group I | Uncomplicated disease, with or without pulmonary artery involvement |
| Group IIA | Mild/moderate single complication together with uncomplicated disease |
| Group IIB | Severe single complication together with uncomplicated disease |
| Group III | Two or more complications together with uncomplicated disease |
Table 4:Ishikawa diagnostic criteria of takayasu's disease40
| Criteria | Definition |
|---|---|
|
Obligatory criterion: Age ≤40 years |
Age 540 yr at diagnosis or at creel of "characteristic signs and symptoms"' of 1 month duration in patient history. |
|
Two major criteria: 1) Left mid suhclavian artery lesion |
The most severe stenosis or occlusion present in the mid portion from the point 1 cm proximal to the left vertebral artery orifice to that 3 cm distal to the orifice determined by angiography. |
| 2) Right mid subclavian artery lesion | The most severe stenosis or occlusion present in the mid portion from the right vertebral artery orifice to the point 3 cm distal to the orifice determined by angiography. |
|
Nine minor criteria 1) High ESR |
Unexplained persistent high ESR ≤20 mm/h (Westergren) at diagnosis or presence of the evidence in patient history. |
| 2) Carotid artery tenderness | Unilateral or bilateral tenderness of common carotid arteries by physician palpation : neck muscle tenderness is unacceptable. |
| 3) Hypertension | Persistent blood pressure ≥140/90 mmHg brachial or ≥160/90 mmHg popliteal at age≤ 40 years, or presence of the history at age ≤40 years |
| 4) Aortic regurgitation or Annuloaortic ectasi | By auscultation or Doppler echocardiography or angiography. By angiography or two-dimensional echocardiography |
| 5) Pulmonary artery lesion | Lobar or segmental arterial occlusion or equivalent determined by angiography or perfusion scintigraphy; or presence of stenosis, aneurysm, luminal irregularity or any combination in pulmonary trunk or in unilateral or bilateral pulmonary arteries determined by angiography |
| 6) Left mid common carotid lesion | Presence of the most severe stenosis or occlusion in the mid portion of 5 cm in length from the point 2 cm distal to its orifice determined by angiography. |
| 7) Distal brachiocephalic trunk lesion | Presence of the most severe stenosis or occlusion in the distal third determined by angiography |
| 8) Descending thoracic aorta lesion | Narrowing, dilation or aneurysm, luminal irregularity or any combination determined by angiography; tortuosity alone is unacceptable. |
| 9) Abdominal aorta lesion | Narrowing, dilation or aneurysm, luminal irregularity or any combination and absence of lesion in aorto-iliac region consisting of 2 cm of terminal aorta and bilateral common iliac arteries determined by angiography; turtuosity alone is unacceptable. |
Management
Medical treatment
Medical treatment is to control active inflammation and minimize arterial injury. To prevent the development of vascular complications and induce remission, early initiation of immunosuppressive treatment is crucial41. Prednisolone is the first line agent, and the EULAR (European League Against Rheumatism) guidelines recommend an initial dose of 1mg/kg/day (total maximum dose 60mg/day), with gradual tapering. Adjunctive steroidsparing immunosuppression is required in the majority of patients to minimise steroid-related complications and control disease progression, particularly as there is considerable risk of relapse when steroid treatment is stopped. There are studies suggesting methotrexate and azathioprine are effective at inducing remission and halting progress of arterial lesions42. A recent review published of 84 patients with TA treated with tumour necrosis factor-? antagonists including infliximab and etanercept revealed complete remission in 37%, partial remission in 53.5%, and 9.5% nonresponders. Of note, side effects (mainly infections and hypersensitivity reactions) were observed in 20% of cases. Likewise, anti-IL-6 receptor monoclonal antibody tocilizumab may help control refractory disease and the published cases have recently been reviewed in the literature43.
Surgical treatment
With symptomatic stenotic or occlusive lesions, it appears appropriate and often necessary to revascularize. The indications for considering intervention include uncontrolled hypertension as a consequence of renal artery stenosis, severe symptomatic coronary artery or cerebrovascular disease, severe aortic regurgitation or coarctation, stenotic or occlusive lesions resulting in critical limb ischemia, and aneurysms at risk of rupture. In these cases the risk benefit ratio for surgery is good44.
Conclusion
• Takayasu’s arteritis is a nonspecific inflammatory disease of the arteries which is more commonly seen in Asian countries and predominantly affects women. Manifestations in our study range from asymptomatic disease, impalpable pulses or arterial bruits ,to catastrophic neurological impairment .
• Disease presentation varies between different populations.
• Angiography remains the gold standard and was helpful in confirming the diagnosis and planning the treatment .Comprehensive angiographic evaluation followed by percutaneous angioplasty and stenting is useful in properly selected cases .
• The four most important complications for classification are Takayasu retinopathy, secondary hypertension, aortic regurgitation, and aneurysm formation, each being graded as mild/moderate or severe at the time of diagnosis.
• Treatment should aim to control disease activity and preserve vascular competence, with minimal long term side effects; those with disease that carries a good prognosis should not be put at risk by treatment that is more harmful than the disease itself.
References
- Kerr GS, Hallahan CW, Giordano J et al. Takayasu arteritis. Ann Intern Med. 1994; 120: 919_29.
- Brunner J, Feldman BM, Pascal N et al. Takayasu arteritis in children and adolescents. Rheumatology. 2010; 49: 1806_14.
- B?c akc ?gil M, Aksu K, Kamal? S et al. Takayasu’s arteritis in Turkey clinical and angiographic features of 248 patients. Clin Exp Rheumatol. 2009; 27(Suppl 52): S59_64.
- Watts R, Al-Taiar A, Mooney J et al. The epidemiology of Takayasu arteritis in the UK. Rheumatology. 2009; 48: 1008_11.
- Saruhan Direskeneli G, Hughes T, Aksu K et al. Identification of multiple genetic susceptibility loci in Takayasu arteritis. Am J Hum Genet. 2013, Advance Access published 2 July 2013, doi:10.1016/j.ajhg. 2013.05.026.
- Sahin Z, Bicakcigil M, Aksu K et al. Takayasu’s arteritis is associated with HLA-B*52 but not with HLA-B*51 in Turkey. Arthritis Res Ther. 2012; 14: R27.
- Numano F, Kakuta T. Takayasu arteritis five doctors in the history of Takayasu arteritis. Int J Cardiol. 1996; 54: S1–10.
- Numano F, Okawara M, Inomata H, et al. Takayasu’s arteritis. Lancet. 2000; 356: 1023–5.
- Shimizu K, Sano K. Pulseless disease. Journal of Neuropathology and Clinical Neurology. 1951; 1.37–1.47.
- Hall S, Barr W, Lie JT, et al. Takayasu arteritis A study of 32 North American patients. Medicine. 1985; 64: 89–99.
- Gravanis MB. Giant cell arteritis and Takayasu aortitis morphologic pathogenetic and etiologic factors. Int J Cardiol. 2000; 75: S21–33.
- Michel BA, Arend WP, Hunder GG. Clinical differentiation between giant cell temporal arteritis and Takayasu’s arteritis. J Rheumatol. 1996; 23: 106–11.
- Boon NA, Colledge NR, Walker BR, et al. Davidson’s principles & practice of medicine. 20th ed. India: Elsevier; 2009.
- Arnaud L, Haroche j, Mathian A, et al. Pathogenesis of Takayasu's arteritis a 2011 update. Autoimm Rev. 2011; 11(1): 61-7.
- Vidhate M, Garg RK, Yadav R, et al. An unusual case of Takayasu's arteritis Evaluation by CT angiography. Ann Indian Acad Neurol. 2011; 14(4): 304-6.
- Kwon HW, Suh YJ, Bang JS, et al. Aortic valve replacement surgery for a case of infantile Takayasu arteritis. Korean J Pediatr. 2012; 55(7): 254-8.
- Gulati A, Bagga A. Large vessel vasculitis. Pediatr Nephrol.2012; 25(6): 1037-48.
- Brady J, Esrig BC, Hamirani K, et al. Severe chronic aortic insufficiency requiring valve replacement an infrequent complication of Takayasu's disease. Echocardiography. 2006; 23(6): 495-8.
- Kumar V, Abbas AK, Fausto N. Robbins and Cotran pathologic basis of disease. 7thed. New Delhi: Thomson Press; 2007.
- Lupi-Herrera E, Sánchez-Torres G, Marcushamer J, et al. Takayasu arteritis Clinical study of 107 cases. Am Heart J. 1977; 93: 94–103.
- Hall S, Barr W, Lie JT, et al. Takayasu arteritis A study of 32 North American patients. Medicine. 1985; 64: 89–99.
- Jain S, Sharma N, Singh S, et al. Takayasu arteritis in children and young Indians. Int J Cardiol. 2000; 75: S153–7.
- Ishikawa K. Natural history and classification of occlusive thromboaortopathy Takayasu’s disease. Circulation. 1978; 57: 27–35.
- Subramanyan R, Joy J, Balakrishnan KG. Natural history of aortoarteritis Takayasu’s disease. Circulation. 1989; 80: 429–37.
- Sharma S, Kamalakar T, Rajani M, et al. The incidence and patterns of pulmonary artery involvement in Takayasu’s arteritis. Clin Radiol. 1990; 42: 177–82.
- Kerr GS, Hallahan CW, Giordano J, et al. Takayasu arteritis. Ann Intern Med. 1994; 120: 919-29.
- Hoffman GS, Ahmed AE. Surrogate markers of disease activity in patients with Takayasu arteritis a preliminary report from The International Network for the Study of the Systemic Vasculitides (INSSYS). Int J Cardiol. 1998; 66 (Suppl 1): S191-S194.
- Matsuyama A, Sakai N, Ishigami M, et al. Matrix metalloproteinases as novel disease markers in Takayasu arteritis. Circulation. 2003; 108: 1469-73.
- Park JH, Han MC, Kim SH, et al. Takayasu arteritis angiographic finding and results of angioplasty. AJR Am J Roentgenol. 1989; 153: 1069-74.
- Yamato M, Lecky JW, Hiramatsu K, et al. Takayasu arteritis radiographic and angiographic findings in 59 patients. Radiology. 1986; 161: 329-34.
- Peachell MB, Muller NL. Pulmonary vasculitis. Semin Respir Crit Care Med. 2004; 25: 483-9.
- Park JH, Chung JW, Lee KW, et al. CT angiography of Takayasu arteritis comparison with conventional angiography. J Vasc Interv Radiol. 1997; 8: 393-400.
- Tanigawa K, Eguchi K, Kitamura Y, et al. Magnetic resonance imaging detection of aortic and pulmonary artery wall thickening in the acute stage of Takayasu arteritis Improvement of clinical and radiologic findings after steroid therapy. Arthritis Rheum. 1992; 35: 476-80.
- Andrews J, Al-Nahhas A, Pennell DJ, et al. Non-invasive imaging in the diagnosis and management of Takayasu’s arteritis. Ann Rheum Dis. 2004; 63: 995-1000.
- Taylor KJ. Arterial vascular ultrasonography. Radiol Clin North Am. 1992; 30: 865-78.
- Lupi-Herrera E, Sánchez-Torres G, Marcushamer J, et al. Takayasu arteritis Clinical study of 107 cases. Am Heart J. 1977; 93: 94–103.
- Arend WP, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990; 33: 1129–34.
- Moriwaki R, Noda M, Yajima M, et al. Clinical manifestations of Takayasu arteritis in India and Japan new classification of angiographic findings. Angiology. 1997; 48: 369–79.
- Ishikawa K. Natural history and classification of occlusive thromboaortopathy Takayasu’s disease. Circulation. 1978; 57: 27–35.
- Ishikawa K. Diagnostic approach and proposed criteria for the clinical diagnosis of Takayasu’s arteriopathy. J Am Coll Cardiol. 1988; 12: 964-72.
- K. Maksimowicz-McKinnon, Clark TM, Hoffman GS. Limitations of therapy and a guarded prognosis in an American cohort of Takayasu arteritis patients. Arthritis and Rheumatism. 2007; vol. 56, no. 3: pp. 1000–1009.
- Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for themanagement of large vessel vasculitis. Annals of the Rheumatic Diseases. 2009; vol. 68, no. 3: pp. 318–323.
- S. Unizony JH Stone, Stone JR. New treatment strategies in large-vessel vasculitis. Current Opinion in Rheumatology. 2013; vol. 25, no. 1: pp. 3–9.
- Fields CE, Bower TC, Cooper LT, et al. Takayasu’s arteritis operative results and influence of disease activity. Journal of Vascular Surgery. 2006; vol. 43, no. 1: pp. 64–71.