
The Known Unknowns: Missing Pieces in in vivo Models of Fragile X Syndrome
Pushkar Kulkarni*, Aarti Sevilimedu*
Centre for Innovation in Molecular and Pharmaceutical Sciences (CIMPS), Dr. Reddy's Institute of Life Sciences, University of Hyderabad Campus, Gachibowli, Hyderabad, Telangana, 500046, India
Abstract
Fragile X Syndrome (FXS) is a rare disease and the leading monogenic cause of Autism Spectrum Disorders (ASD). It is caused by the silencing of the Fragile X mental retardation (FMR1) gene and the subsequent reduction or loss of fragile X mental retardation protein (FMRP). The clinical effects seen in FXS patients are several and highly variable making it difficult to model them in a single model or even one organism. Furthermore, several human behaviours can be measured only through surrogate endpoints in animals. Therefore, it has been challenging to develop in vivo models of FXS for drug discovery.
This review endeavours to consolidate the information on all available in vivo models for FXS specifically with a focus on their suitability for drug development, with the objective of identifying gaps and potential solutions. To do so, we have summarised the major clinical characteristics and possible mechanisms underlying clinical phenotypes associated with FXS and other disorders that arise from abnormalities in the FMR1 locus, such as fragile-X associated tremor/ataxia syndrome (FXTAS), fragile-X-associated neuropsychiatric disorders (FXAND) including ASD and fragile x-associated primary ovarian insufficiency (FXPOI). We then connect clinical features to phenotypes observed in available in vivo FXS models where possible, covering a wide range of organisms from primates, rats, mice, zebrafish, fruit fly and zebra finches. For each model organism, we list the technology of model creation, phenotypes/assays, mechanistic basis of disease manifestation and specific advantages or disadvantages of the model in the context of drug discovery.
Finally, we have highlighted the missing pieces in FXS modelling and propose strategies to address them, considering aspects of modelling spectrum disorders, repeat expansion and silencing, new functions of FMRP and identification of efficacious treatments.
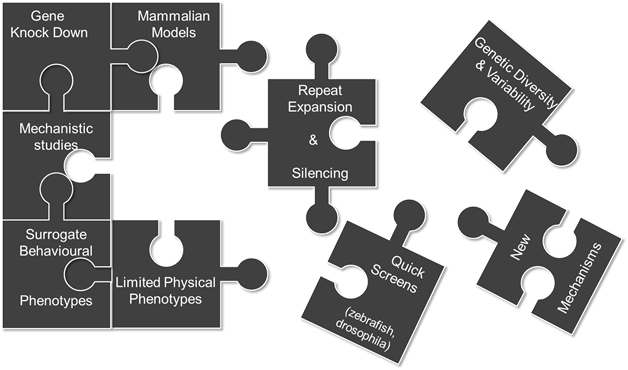
Figure: Missing pieces in modelling of Fragile X Syndrome
Introduction
Fragile X Syndrome (FXS) is one of the most studied monogenic neurological syndromes over the past few decades. It is caused by silencing of the Fragile X mental retardation (FMR1) gene and the subsequent reduction or loss of FMR protein (FMRP). CGG repeat expansion in the 5’UTR of the FMR1 gene, followed by hypermethylation of the region is the basis of the observed silencing in FXS1. This event occurs sequentially in successive generations, beginning with small repeat expansion (55-200) causing pre-mutation in one generation with a toxic gain of function in the mRNA, followed by further expansion (>200) to full mutation in subsequent generations with complete silencing of the gene2. FMRP is an RNA binding protein3, a well-known regulator of translation, and is known to interact with well over 800mRNAs in the adult neuron4. Even though several of the mRNA targets have not been fully characterised, it can be said that, FMRP loss has a cascading effect on several pathways which result in the observed clinical features5,6. Several model organisms have been used to model this disease, as discussed extensively in the following tables. The major challenges in modelling FXS are listed here (and substantiated in the rest of the review article):
a) The clinical presentation of the disease in humans is highly variable with different individuals showing different sets of clinical phenotypes.
b) Similar to other neurological diseases, the biochemical and pathological profiling of FXS in real time is practically impossible. This makes it extremely difficult to decipher the human pathobiology making it necessary to have in vivo models.
c) The human nervous system is the most evolved and mimicking it in lower mammals and other vertebrates is done only by using surrogate endpoints especially for cognitive and social behaviours.
d) Animal models show high variability in measurable phenotypes.
Therefore, modelling a complex neurological syndrome like FXS is a continuing process and will require a set of organisms to model all the clinical characteristics, to study various pathobiological aspects and to screen potential drug candidates.
Modelling most diseases and syndromes is necessary in disciplines of disease biology and drug discovery; however, given FXS’s monogenic aetiology, these models can be potentially studied for understanding several other aspects including (but not limited to):
a) Pathways of development of various cognitive abilities
b) Specific connective tissue organogenesis
c) Pathways of behaviours like anxiety, depression, irritability, others.
Thus, modelling FXS and improving the models can have great value to the field of neurosciences. In the present review, we discuss clinical presentations and possible mechanisms, various in vivo models and their advantages and disadvantages, and propose the next set of models and methods that can be used to plug in the missing pieces.
Clinical characteristics of Fragile X Syndrome and the suggested mechanisms
Clinical characteristics that are most commonly noticed in patients with FXS and the suggested mechanisms have been presented below in Table 1.
Table 1: COMMON CLINICAL CHARACTERISTICS AND THEIR SUGGESTED MECHNANISMS
|
Clinical Presentations |
Suggested Mechanisms of Pathobiology |
Reference/s |
|
Fragile X-associated neuropsychiatric disorders (FXAND) Attention-Deficit Hyperactivity Disorder (ADHD) Anxiety Autism Spectrum Disorder (ASD) Developmental Intellectual Disability Language Deficits Seizures (rarely) |
Dysregulation of excitatory and inhibitory neurotransmission by:
Indirect glutamatergic mechanisms that modulate mGluR:
|
[5]–[8] |
|
|
|
|
|
Fragile-X associated tremor/ataxia syndrome (FXTAS) |
Excess mRNA from FMR1 premutation may lead to FXTAS in the progeny through the following mechanisms:
Its predicted that CCG repeats may lead to sequestration of specific RNA-binding proteins such as lamin A/C, Pura, hnRNP, Sam68, and drosha, thus affecting some of the normal cell function in both FXTAS and FXPOI. |
[9], [10]
|
|
|
|
|
|
Fragile X-Associated Primary Ovarian Insufficiency (FXPOI) |
Repeat-associated non-AUG (RAN)-translated poly-glycine species (FMR polyG) leading to intracellular inclusions bodies affecting ovarian function. There is an overlap of mechanisms suggested for FXTAS and FXPOI. |
[11], [12]
|
|
|
|
|
|
Spine abnormalities |
FMRP loss-related dendritic abnormalities due to the following:
|
[5], [13], [14] |
|
|
|
|
|
Macroorchidism |
Increase in hydrophilic glycoprotein granules causing testicular edema followed by scattered collagen fibres and bundles of microfibril. Exact pathway has not been elucidated but may be attributed to connective tissue dysregulation. |
[15], [16] |
|
|
|
|
|
Other Physical Characteristics Macrocephaly Long Face Prominent Ears Prominent Jaw Flat Feet Joint Hypermobility |
Loss of FMRP can lead to dysregulation of the following extracellular matrix and connective tissue components:
|
[16], [5] |
In vivo models of Fragile X Syndrome
Various in vivo models of FXS have been described in Table 2. We have also provided the potential advantages and disadvantages of each of these models.
Table 2: DESCRIPTION OF VARIOUS IN VIVO MODELS OF FXS AND THEIR ADVANTAGES AND DISADVANTAGES
|
Species, Strain, Model |
Technology used to create model |
Phenotypes |
Mechanisms |
Reference/s |
Advantages |
Disadvantages |
|
Primate |
|
|||||
|
Non-human primate, Macaca mulatta, Spontaneous |
None, analysis from a population of macaque |
Not measured |
Not evaluated. FMR1 alleles carried out to assess CAG/CGG repeat distribution |
[17] |
CGG repeat distribution and propensity for expansion most similar to that in humans; |
Instance of FXS was not observed; |
|
Rat |
|
|||||
|
Sprague Dawley, Fmr1 em1Sage |
Zinc-finger-nuclease mediated knockout of Fmr1 (exon 8) in SD or LEH background |
Macroorchidism, alterations in density of dendritic spines; Impaired associative learning and memory. |
Loss of FMRP, altered translation profile and development of prefrontal cortex circuitry, altered mGluR5 signalling |
[18]–[21] |
Allows: |
Rat models do not offer significant advantages over murine models with regard to behavioural phenotypes of FXS. |
|
LEH, Fmr1em1/PWC |
Zinc-finger-nuclease mediated knockout of Fmr1 in SD or LEH background |
|
||||
|
Sprague Dawley, Fmr1 exon4 KO |
CRISPR-Cas9, exon 4 mutation leading to truncation |
Macroorchidism, altered LTP, LTD, spatial learning and memory; impaired social interaction (novelty recognition) |
Loss of FMRP, altered translation and synaptic plasticity |
[22] |
Clear evidence of deficits in hippocampus-dependent spatial learning and memory (unlike murine models) |
|
|
Mouse |
|
|||||
|
Knock-outs: C57BL6/J or FVB/N, Sighted FVB, Fmr1tm1Cgr |
Homologous recombination with disruption in exon 5 |
Macroorchidism; |
Loss of FMRP, altered translation profile and altered mGluR5 signalling |
[23]–[25] and others, summarized in [26]
|
Most commonly used model; |
Residual Fmr1 mRNA present, repeat expansion and promoter methylation absent; |
|
Conditional KOs: Sighted FVB or C57BL/6J, Fmr1 cON, CKO, KO2 |
Cre-lox recombination with disruptions in exon/intron 1 |
[27] |
Allows tissue or brain region specific function of FMRP (such as in the hippocampus, CA1 neurons etc) as well as temporal regulation (as yet unreported). |
Not very widely used, offers no specific advantages over the KO models for general behavioural tests. |
||
|
Point mutation: C57BL6/J and FVB/N, Fmr1I304N |
Cre-lox recombination |
Phenocopies KO models |
Missense mutation in the KH domain of FMRP prevents RNA as well as ribosome binding, leads to reduced FMRP levels. |
[28] |
Additional model with a different genotype, same phenotype, for drug discovery; |
Models a very rarely observed patient mutation. |
|
Rescue models: C57BL6/J Fmr1 or Fmr1KO TG298, Fmr1 TG296 |
YAC transgenesis of human FMR1alleles with 20/92 repeats, into WT |
Altered anxiety related phenotypes contrary to those observed in FXS KO |
Overexpression of FMRP (~10 fold over endogenous); |
[29] |
Reveals phenotypes associated with FMRP overexpression; |
No major repeat expansion observed; |
|
YAC transgenesis of human FMR1alleles with 20/92 repeats, into Fmr1 KO mice |
Rescue of macroorchidism, and anxiety related phenotypes |
Rescue of FMRP deficit, with overexpression of FMRP; |
[30] |
|||
|
Knock In/Repeat Expansion C57BL6/J or FVB/N, Fmr1tm2Cgr |
Homologous recombination |
Impaired locomotion, altered anxiety, spatial memory and learning deficits |
Moderate repeat expansion and instability, leading to reduced FMRP and increased Fmr1 mRNA, intranuclear inclusions in the neurons and astrocytes |
Summarized in [31] |
Only model to study mechanisms of pathogenic repeat expansion similar to that observed in humans with FXS premutation |
Repeat expansion is not accompanied by promoter methylation and silencing, even though FMRP level is reduced; |
|
Zebrafish |
|
|||||
|
Knockdown (transient) |
Morpholino |
Morphological |
Reduction in Fmrp, increased mGluR5, perturbed calcium distribution and signalling |
[32] |
Simple, robust knockdown produces strong morphological changes; |
Morpholino based induction of artefacts may confound some findings; |
|
fmr1hu2787 (stop); fmr1hu2898 (splice) |
ENU, (N-ethyl-N-nitrosourea) induced mutations and screening for fmr1knock out |
No anatomical defects. |
Loss of Fmrp, increased mGLuR5 |
[33]–[35], |
Loss of function model illustrates possible mechanism. Demonstrates social deficits. |
Mild phenotypes, not suitable for screening. Potential compensatory mechanisms in play. |
|
fmr1-/- mutant |
CRISPR/Cas9 |
Craniofacial abnormalities, Hyperactivity, Memory deficit |
Loss of Fmrp, changes in the expression of genes involved in learning and memory and cartilage development. |
[36] |
First targeted knockout model showing phenotypes similar to observed human FXS symptoms. |
Mild phenotypes are observed, potentially due to compensatory mechanisms; |
|
Knock down (transient) |
DNAzyme |
Craniofacial abnormalities, |
Reduction in Fmrp, increased mGLuR5, altered p-ERK and p-Akt |
[37], [38] |
Simple, inexpensive transient model for drug screening; |
Transient knock down. Social deficits cannot be appreciated in short duration knock down. |
|
Drosophila |
|
|||||
|
dfmr1 mutant |
P-element insertion |
Abnormalities in behaviour, including erratic activity, disrupted sleep patterns, circadian clock; defects in synaptogenesis and spermatogenesis; defects in learning and memory (olfactory, courtship assays) |
Loss of Fmrp, impacting glutamate signalling (mGluR5) DNA damage response (DDR), RNA editing (ADAR) and microRNA pathways (Ago1) |
[39], [40] |
Simple model recapitulating various human and mouse FXS phenotypes; |
Promoter methylation and silencing is not modelled; |
|
Round worm, Caenorhabditis Elegans pAWC::FMR(CGG)99 |
Expression of varying CGG repeat length containing reporters. |
Loss of olfactory adaptation due to a repeat-associated dysfunction |
Repeat containing RNA acts through miRNA-binding Argonaute ALG-2, to impact adaptive olfactory response (a readout for reduced neuronal plasticity) |
[41] |
Potential model to study olfactory plasticity and subversion in neuronal function, and investigate the molecular link between expanded repeats and plasticity in a simplified system. |
Only one published report of this model. The reproducibility of this model needs to be established. |
|
Zebra finch, Taeniopyia guttata |
No model yet |
Vocalization & Language development is dependent on fmr1 expression. |
Mechanisms unknown |
[42], [43] |
Potential model to study the mechanisms behind speech and language pathology. |
No model developed so far, feasibility unknown. |
Missing pieces in modelling of FXS and potential solutions
Modelling spectrum disorders
Fragile X syndrome is the leading genetic cause of autism, and because of the implication of a single causal gene, animal models of FXS are numerous26. The characteristic clinical manifestation of FXS involves some or all of the following symptoms: long face, macrocephaly, prominent ears, prominent jaw, flat feet, joint hypermobility, macroorchidism (clinical); attention-deficit hyperactivity disorder (ADHD), anxiety autism spectrum disorder (ASD) (psychological), developmental intellectual disability, language deficits (developmental) and strabismus, recurrent otitis, gastrointestinal complaints, obesity and seizures (less prevalent)44. While the various models capture a subset of these phenotypes (see table 2 above), no single model has been able to mimic the spectrum of symptoms and deficits seen in human FXS patients, and this has severely impacted screening and drug discovery efforts. There could be two potential reasons for this:
(i) The first is that the presence, severity and manifestation of FXS symptoms varies widely even in human patients44, and is likely influenced strongly by the genetic background and environmental factors. Therefore, one can argue that inconsistencies are expected in the models as well.
(ii) The second stems from the nature and function of the FMR protein. FMRP is a regulator of translation and is expressed from very early on during development, which means that when FMRP is silenced, the levels of a number of proteins (several hundred in an adult neuron, for example), and consequently a number of signalling pathways are likely to be altered, and differentially so at various stages during development45. The phenotypes observed are a result of these cumulative changes. Even though FMRP per se is fairly well conserved in all of the models, varying degrees of conservation across the target proteins, pathways and differentiation paradigms are likely to cause inconsistencies in phenotypes observed in the models. Restricting discovery programs to one model organism, or picking a single genetic background in a model, is unlikely to be beneficial, since heterogeneity is a feature of the disease, not the model. Therefore, we would like to propose that diversification, both in terms of model organisms and genetic backgrounds may be key to identifying robust and efficacious therapeutic interventions. Focussing on simpler genetic models like Zebrafish and Drosophila, may allow one to conduct high throughput screens in a genetically diverse population, and generate statistically significant results.
Repeat expansion and silencing
In FXS, a repeat expansion in the 5’ UTR of the human FMR1 gene leads to hypermethylation and silencing, which results in a drastic reduction of FMRP. Therefore, control at the level of repeat expansion or methylation are the best therapeutic avenues. In humans, the FMR1 locus naturally contains 6-55 CGG repeats; however, all of the model organisms (except the primate) contain very few repeats, if any. The mechanism of repeat expansion involves different stages in which the premutation stage (55-200 repeats) is associated with an increase in FMR1 RNA levels (toxic gain of function), which may be a prerequisite for the progression to full expansion (>200 repeats) leading to hypermethylation and subsequent silencing. Therefore, lack of a critical number of repeats at the outset, combined with a lack of progressive expansion could explain why the knock-in models do not show any promoter methylation-dependent silencing. With a carefully chosen combination of artificial repeat insertion, control of FMR1 RNA levels and manipulation of the DNA replication or repair pathways to promote slippage, it may be possible to develop such a model. We believe that such studies can be conducted in simpler models like yeast46, used to derive optimal conditions which may then be moved into higher models like Zebrafish and mouse, to create true FXS models. Interventions which aim to interfere with repeat expansion, or those that revert the hypermethylation-driven silencing (small molecules47-49, or genetic interventions50,51) can then be screened in such a model, and will either significantly ameliorate the disease (even a two-fold increase in FMRP level is associated with a significantly higher IQ52) or better still, prevent it.
New functions of FMRP
Since FXS is primarily seen as a disease of the brain, studies have focussed on FMRP’s neuronal function and neuronal phenotypes in the models. FMRP is ubiquitously expressed during early developmental stages, and it is increasingly clear that FMRP interacts with and regulates diverse cellular pathways in addition to its primary function as a translation regulator in neurons. These include regulation of RNA editing and splicing, chromatin structure, cellular differentiation kinetics, ion channel regulation and microRNA pathways53. Therefore, exploring the molecular mechanisms underlying the contribution of these pathways to disease progression or phenotypes could be an important new avenue of research. Traditional translation targets of FMRP may also be influenced by disruptions in these other pathways (for example, AMPA receptor and RNA editing54, microRNA and ion channels55) and may be better rescued by novel treatments or combination therapies.
Identifying efficacious treatments for FXS- moving the needle on the preclinical side
While molecular mechanisms of FXS are fairly well understood, and small molecules targeting at least ten different pathways are able to rescue several molecular phenotypes such as protein synthesis, synaptic plasticity and calcium regulation, none of these have translated into clinical efficacy in humans56. While one obvious explanation is that this is due to the complexity of, and our lack of understanding of the human system, there are alternative explanations which require due consideration. An important take-away from the clinical trials conducted so far, is that tests which measure core phenotypes like behaviour and cognition directly and not through surrogate indications, need to be developed in order to determine the true efficacy of drug treatment. However, there are many avenues for improvement on the preclinical side.
(i) First, newer models which capture the repeat expansion and methylation features should be developed, since targeting these upstream nodes will result in maximal impact (section above).
(ii) Second, varied genetic backgrounds and multiple model organisms should be employed in order to determine the robustness of the phenotypes or treatment being assessed. The molecular phenotypes are highly conserved from flies to human, and the small molecules being considered for clinical trials have been identified based on these conserved pathways. It may be prudent therefore to conduct such studies in models like Danio rerio (Zebrafish) where true “wild type” animals (wild caught) can be used (incorporating the genetic diversity present in the natural world), with as large a sample size as required to power the statistics. Such an approach may allow one to incorporate the varied baselines in the population (for example, the median increase in anxiety in wild-caught vs. lab-wild type strains50) and multi-factorial influences on the neurological phenotypes during the screening process to make results more robust57. Techniques for simple, rapid and inexpensive model creation, such as transient knockdown of gene expression (using DNAzymes or morpholinos in zebrafish32,37,58, RNAi in D.melanogaster59, and differentiation of patient derived cell lines60) will also aid in increasing the number of varied models available to assess the same phenotype and its treatment. A larger number of treatments (compounds and paradigms) can be tested in a more diverse set of assays in such models, and may drastically improve the chances of finding a drug that will translate well into humans compared to traditional approaches using the mouse model.
(iii) Third, multiple tests or assays which measure the same parameter should be employed in each study, and at least one of these should measure the same parameter as in a clinical trial (such as fMRI or EEG).
(iv) Fourth, drug screening should be conducted in models where the link between the molecular changes due to FMRP loss, to circuitry and behaviour are well-established (such as in olfactory system of D.melanogaster61), and rescue at each stage should be assessed.
(v) Fifth, a number of treatment windows may need to be explored for each class of drug, coupled with longitudinal studies which measure impact over the long term, especially for treatments targeting behaviour. Given that the circuitry can be modulated only in certain windows during development, earlier treatment windows need to be preferentially identified and studied.
(vi) Finally, FMRP appears to play a role in multiple unconnected pathways, therefore genetic studies in models like flies and zebrafish could be used to better understand these new molecular functions of FMRP. Subsequently, combination treatments to address more than a single target at a time may be prioritized for screening.
Disruptive platforms and niche models
Given that decades of therapeutic research using the available models of FXS have not led to the identification of a clinically efficacious drug, the possibility that the molecular landscape and regulatory network in the human brain is not sufficiently or completely replicated in any other model, has to be considered for the next phase of therapeutic research. In such a scenario, critical and validated endophenotypes62 may need to be used as a basis for screening directly in a “human” model. Brain organoids satisfy the need for “human origin” as well as provide the genetic, cellular and architectural features of the human brain and could therefore be a powerful platform for identification of critical endophenotypes as well as for screening63. Brain organoids from patient derived cells (hiPSCs) have been used to study varied diseases of the central nervous system64, and are likely to be relevant to FXS, where phenotypes are thought to stem from defective cross-talk between multiple cell types and altered neural circuitry. However, the current state of this technology in terms of the time, skill and expense involved, as well as the inability to sustain organoids in culture to model adult-phenotypes limits its applicability to drug discovery, as on date. Yet another strategy to conduct studies on human origin brain tissue, in an in vivo setting is the clever use of transplanted FXS brain tissue (iPSCs which differentiate after transplantation or neural precursor cells (NPCs)) into the mouse brain65. While the chimeric setting has the same limitations as the mouse model in terms of screening for behavioural or cognitive end points, it is likely to reveal the most authentic, cell-type and microenvironment specific responses to drugs.
Conclusions and future outlook
Fragile X syndrome is a classic test case for a rare disease model, which despite the availability of numerous models and studies over the decades, has not yielded any therapeutic benefits for patients. We believe that part of the reason for this has been the use of approaches which were developed and standardized on the basis of what has worked for the more prevalent, non-monogenic diseases that have dominated the clinical research and drug development fields. In the case of drug development for rare monogenic disorders where disease manifestation is heavily influenced by the underlying genetic background and treatment needs are primarily symptomatic, radically different approaches like the ones described above may be more fruitful. Implementation of such strategies and the consequent identification of efficacious treatments may cause a paradigm shift in the way rare disease biology, modelling and drug development is practiced in the future.
Funding and Acknowledgements
Work in the authors’ laboratories is supported by funding from Dr.Reddy’s Institute of Life Sciences, Hyderabad, India and Department of Biotechnology (DBT) grant BT/PR28305/GET/119/272/2018 awarded to PK and AS. The authors would like to acknowledge Dr. K. Chatti and Dr. K. Parsa for valuable comments on the manuscript.
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Intellectual Property Statement: The method for creating zebrafish knock down models using DNAzymes is the intellectual property of Dr. Reddy’s Institute of Life Sciences and is covered by the following patent: Embryonic Zebrafish models using DNAzyme mediated knockdown. Sevilimedu A, Kulkarni P. Application No. PCT/IB2019/051255, Indian Application No. 01741031477. Complete specification filed for Dr. Reddy’s Institute of Life Sciences. The authors are affiliated to Dr. Reddy’s Institute of Life Sciences.
References
- Oberle I, Rousseau F, Heitz D, et al. “Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome,” Science. 1991; 80252(5009): 1097–1102, doi: 10.1126/science.252.5009.1097.
- Fu YH, Kuhl DP, Pizzuti A, et al., “Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox,”. Cell. Dec 1991; 67(6): 047–1058 , doi: 10.1016/0092-8674(91)90283-5.
- Ashley CT, Wilkinson KD, Reines D, et al. “FMR1 protein: Conserved RNP family domains and selective RNA binding”. Science. 1993; 80262(5133): 563–566, doi: 10.1126/science.7692601.
- Darnell JC, Driesche SJV, Zhang C, et al., “FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism,”. Cell. 2011; 146(2): 247–261, doi: 10.1016/j.cell.2011.06.013.
- Salcedo-Arellano MJ, Dufour B, McLennan Y, et al. “Fragile X syndrome and associated disorders: Clinical aspects and pathology,”. Neurobiology of Disease. Mar, 01, 2020; 136. Academic Press Inc p. 104740, , doi: 10.1016/j.nbd.2020.104740.
- Hagerman RJ, Berry-Kravis E, Hazlett HC, et al. “Fragile X syndrome,” Nature reviews. Disease primers. Sep 29, 2017; 3(1). Nature Publishing Group, p. 17065, , doi: 10.1038/nrdp.2017.65.
- Hugger Toft AK, Lundbye CJ, Banke TG. “Dysregulated NMDA-receptor signaling inhibits long-term depression in a mouse model of fragile X syndrome,”. J Neurosci. Sep 2016; 36(38): 9817–9827, doi: 10.1523/JNEUROSCI.3038-15.2016.
- Cheng GR, Xiang YD, Liu D, et al. “The Implication of AMPA Receptor in Synaptic Plasticity Impairment and Intellectual Disability in Fragile X Syndrome,”. Physiol Res. 2017; 66(715–727), doi: 10.33549/physiolres.933473.
- Giulivi C, Napoli E, Tassone F, et al. “Plasma metabolic profile delineates roles for neurodegeneration, pro-inflammatory damage and mitochondrial dysfunction in the FMR1 premutation”. Biochem J. 2016; 473(21): 3871–3888, doi: 10.1042/BCJ20160585.
- Elizur SE, Dratviman-Storobinsky O, Derech-Haim S, et al., “FMR6 may play a role in the pathogenesis of fragile X-associated premature ovarian insufficiency,”. Gynecol Endocrinol. Apr 2016; 32(4): 334–337, doi: 10.3109/09513590.2015.1116508.
- Fink DA, Nelson LM, Pyeritz R, et al., “Fragile X Associated Primary Ovarian Insufficiency (FXPOI): Case Report and Literature Review,”. Front Genet. Nov 2018; 9: 529, doi: 10.3389/fgene.2018.00529.
- Man L, Lekovich J, Rosenwaks Z, et al “Fragile X-associated diminished ovarian reserve and primary ovarian insufficiency from molecular mechanisms to clinical manifestations,”. Frontiers in Molecular Neuroscience. Sep 12 2017; 10 Frontiers Media SA, doi: 10.3389/fnmol.2017.00290.
- Pasciuto E, Ahmed T, Wahle T, et al., “Dysregulated ADAM10-Mediated Processing of APP during a Critical Time Window Leads to Synaptic Deficits in Fragile X Syndrome,”. Neuron. Jul 2015; 87(2): 82–398, doi: 10.1016/j.neuron.2015.06.032.
- Bilousova TV, Dansie L. M. Davidian, et al. “PRENATAL PATHOGENESIS OF MACROORCHIDISM IN THE FRAGILE X SYNDROME,”. Pediatr Res. Apr 1987; 21(4): 230A-230A, doi: 10.1203/00006450-198704010-00383.
- Shapiro LR, Wilmot PL, Omar RA, et al. “PRENATAL PATHOGENESIS OF MACROORCHIDISM IN THE FRAGILE X SYNDROME,”. Pediatr Res. Apr 1987; 21(4): 230A-230A, doi: 10.1203/00006450-198704010-00383.
- Ramírez-Cheyne JA, Duque GA, Ayala-Zapata S, et al. “Fragile X syndrome and connective tissue dysregulation,”. Clin Genet. Feb 2019; 95(2): 262–267, doi: 10.1111/cge.13469.
- Arocena DG, Breece KE, Hagerman PJ. “Distribution of CGG repeat sizes within the fragile X mental retardation 1 (FMR1) homologue in a non-human primate population,”. Hum Genet. 2003; 113(5): 371–376, doi: 10.1007/s00439-003-0982-9.
- Hamilton SM, Green JR, Veeraragavan S, et al., “Fmr1 and Nlgn3 knockout rats: Novel tools for investigating autism spectrum disorders,”. Behav Neurosci. Apr 2014; 128(2): 103–109, doi: 10.1037/a0035988.
- Till SM, Asiminas A, Jackson AD, et al. “Conserved hippocampal cellular pathophysiology but distinct behavioural deficits in a new rat model of FXS,”. Hum Mol Genet. 2015; 24(21): 5977–5984, doi: 10.1093/hmg/ddv299.
- Berzhanskaya J, Phillips MA, Gorin A, et al. “Disrupted Cortical State Regulation in a Rat Model of Fragile X Syndrome,”. 2016, doi: 10.1093/cercor/bhv331.
- Asiminas A, Jackson AD, Louros SR, et al. “Sustained correction of associative learning deficits after brief, early treatment in a rat model of Fragile X Syndrome,”. Sci Transl Med. 2019; 11( 494): 1–11, doi: 10.1126/scitranslmed.aao0498.
- Tian Y, Yang C, Shang S, et al., “Loss of FMRP impaired hippocampal long-term plasticity and spatial learning in rats,”. Front Mol Neurosci. Aug 2017; 10, doi: 10.3389/fnmol.2017.00269.
- The Dutch-Belgian Fragile X Consortium, “Fmrl Knockout Mice: A Model to Study Fragile X Mental Retardation,”. 1994.
- Yan QJ, Rammal M, Tranfaglia M, et al “Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP,”. Neuropharmacology. Dec 2005; 49(7): 1053–1066, doi: 10.1016/j.neuropharm.2005.06.004.
- Dobkin C, Rabe A, Dumas R, et al. “Fmr1 knockout mouse has a distinctive strain-specific learning impairment,”. Neuroscience. Sep 2000; 100(2): 423–429, doi: 10.1016/S0306-4522(00)00292-X.
- Kooy RF, Jin P, Bao H, et al. “Animal models of fragile X syndrome,” in Fragile X Syndrome: From Genetics to Targeted Treatment Rob Willemsen and R Frank Kooy Ed. Elsevier Inc. 2017; 123–147.
- Mientjes EJ, Nieuwenhuizen I, Kirkpatrick L, et al. “The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo,”. Neurobiol Dis. Mar 2006; 21(3): 549–555, doi: 10.1016/j.nbd.2005.08.019.
- Zang JB, Nosyreva ED, Spencer CM, et al. “A Mouse Model of the Human Fragile X Syndrome I304N Mutation,”. PLoS Genet. Dec 2009; 5(12): e1000758, doi: 10.1371/journal.pgen.1000758.
- Peier AM. “(Over)correction of FMR1 deficiency with YAC transgenics: behavioral and physical features,”. Hum Mol Genet. May 2000; 9(8): 1145–1159, doi: 10.1093/hmg/9.8.1145.
- Peier AM, Nelson DL. “Instability of a premutation-sized CGG repeat in FMR1 YAC transgenic mice,”. Genomics. 2002; 80(4): 423–432, doi: 10.1006/geno.2002.6849.
- Berman RF, Buijsen RA, Usdin K, et al. “Mouse models of the fragile X premutation and fragile X-associated tremor/ataxia syndrome,”. Journal of Neurodevelopmental Disorders. Jan 30 2014; 6(1): 1–16, Springer New York LLC, doi: 10.1186/1866-1955-6-25.
- Tucker B, Richards RI, Lardelli M. “Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis , craniofacial development and fragile X syndrome,”. 2006; 15(23): 3446–3458, doi: 10.1093/hmg/ddl422.
- den Broeder MJ, van der Linde H, Brouwer JR, et al. “Generation and characterization of FMR1 knockout zebrafish”. PLoS One. Jan 2009; 4(11): e7910, doi: 10.1371/journal.pone.0007910.
- Ng MC, Yang YL, Lu KT. “Behavioral and synaptic circuit features in a zebrafish model of fragile X syndrome”. PLoS One. Jan 2013; 8(3): e51456, doi: 10.1371/journal.pone.0051456.
- Wu YJ, Hsu MT, Ng MC, et al., “Fragile X Mental Retardation-1 Knockout Zebrafish Shows Precocious Development in Social Behavior,”. Zebrafish. Oct 2017; 14(5): 438–443, doi: 10.1089/zeb.2017.1446.
- Hu J, Chen L, Yin, J et al. “Hyperactivity, Memory Defects, and Craniofacial Abnormalities in Zebrafish fmr1 Mutant Larvae,”. Behav Genet. 2020; 0123456789, doi: 10.1007/s10519-020-09995-7.
- Medishetti R, Rani R, Kavati S, et al. “A DNAzyme based knockdown model for Fragile-X syndrome in zebrafish reveals a critical window for therapeutic intervention,”. J Pharmacol. Toxicol Methods. November 2019; 101: 106656, 2019, doi: 10.1016/j.snb.2019.127065.
- Sevilimedu A, Kulkarni P. “Embryonic Zebrafish models using DNAzyme mediated knockdown”. 2019. PCT/IB2019/051255.
- Morales J, Hiesinger PR, Schroeder AJ, et al. “Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain”. Neuron. Jun 2002; 34(6): 961–72, doi: 10.1016/s0896-6273(02)00731-6.
- Dockendorff TC, Su HS, McBride SMJ, et al. “Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest,”. Neuron. Jun 2002; 34(6): 973–984, doi: 10.1016/S0896-6273(02)00724-9.
- Juang BT, Ludwig AL, Benedetti KL, et al. “Expression of an expanded CGG-repeat RNA in a single pair of primary sensory neurons impairs olfactory adaptation in Caenorhabditis elegans”. Hum Mol Genet. 2014; 23(18): 4945–4959.
- Winograd C, Clayton D, Ceman S. “Expression of fragile X mental retardation protein within the vocal control system of developing and adult male zebra finches,”. Neuroscience. Nov 2008; 157(1): 132–142, doi: 10.1016/j.neuroscience.2008.09.005.
- Winograd C, Ceman S. “Exploring the zebra finch Taeniopygia guttata as a novel animal model for the speech-language deficit of fragile X syndrome,”. Results Probl Cell Differ. 2012; 54: 181–197, doi: 10.1007/978-3-642-21649-7_10.
- Rajaratnam A, Shergill J, Salcedo-Arellano M, et al. “Fragile X syndrome and fragile X-associated disorders”. F1000Research. 2017; 6: 2112, doi: 10.12688/f1000research.11885.1.
- Santoro MR, Bray SM, Warren ST. “Molecular Mechanisms of Fragile X Syndrome: A Twenty-Year Perspective,”. Annu Rev Pathol Mech Dis. 2012; 7(1): 219–245, doi: 10.1146/annurev-pathol-011811-132457.
- Khristich AN, Mirkin SM. “On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability,”. doi: 10.1074/jbc.REV119.007678.
- Kumari D, Sciascia N, Usdin K. “Small molecules targeting H3K9 methylation prevent silencing of reactivated FMR1 alleles in fragile X syndrome patient derived cells,”. Genes (Basel). Apr 2020; 11(4), doi: 10.3390/genes11040356.
- Chiurazzi P, Pomponi MG, Pietrobono R, et al. “Synergistic effect of histone hyperacetylation and DNA demethylation in the reactivation of the FMR1 gene”. 1999.
- Kumari D, Gazy I, Usdin K. “Pharmacological reactivation of the silenced FMR1 gene as a targeted therapeutic approach for fragile X syndrome,”. Brain Sciences. 2019; 9(2). MDPI AG, doi: 10.3390/brainsci9020039.
- Liu XS, Wu H, Krzisch M, et al. “Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene,”. Cell. Feb 2018; 172(5): 979-992.e6, doi: 10.1016/j.cell.2018.01.012.
- Xie N, Gong H, Suhl JA, et al. “Reactivation of FMR1 by CRISPR/Cas9-Mediated Deletion of the Expanded CGG-Repeat of the Fragile X Chromosome,”. PLoS One. Oct 2016; 11(10): e0165499, , doi: 10.1371/journal.pone.0165499.
- Kim K, Hessl D, Randol JL, et al. “Association between IQ and FMR1 protein (FMRP) across the spectrum of CGG repeat expansions,”. PLoS One. Dec 2019; 14(12): e0226811, doi: 10.1371/journal.pone.0226811.
- Dockendorff TC, Labrador M. “The Fragile X Protein and Genome Function,”. Molecular Neurobiology. Jan 01 2019; 56(1): 711–721. Humana Press Inc, doi: 10.1007/s12035-018-1122-9.
- Shamay-Ramot A, Khermesh K, Porath HT, et al. “Fmrp Interacts with Adar and Regulates RNA Editing, Synaptic Density and Locomotor Activity in Zebrafish,”. PLOS Genet. Dec 2015; 11(12): e1005702, doi: 10.1371/journal.pgen.1005702.
- Loohuis NFMO, Ba W, Stoerchel PH, et al. “MicroRNA-137 Controls AMPA-Receptor-Mediated Transmission and mGluR-Dependent LTD”. Cell Rep. 2015; 11: 1876–1884, doi: 10.1016/j.celrep.2015.05.040.
- Berry-Kravis EM, Lindemann L, Jønch AE, et al. “Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome,”. Nat Rev Drug Discov. Dec 2017, doi: 10.1038/nrd.2017.221.
- Roy T, Bhat A. “Divergences in learning and memory among wild zebrafish: Do sex and body size play a role?,”. Learn Behav. Jun 2018; 46(2): 124–133, doi: 10.3758/s13420-017-0296-8.
- Dwivedi S, Medishetti R, Rani R, et al. “Larval zebrafish model for studying the effects of valproic acid on neurodevelopment: An approach towards modeling autism,”. J Pharmacol. Toxicol Methods. Nov 2019; 95: 56–65, doi: 10.1016/J.VASCN.2018.11.006.
- Drozd M, Bardoni B, Capovilla M. “Modeling fragile X syndrome in drosophila,”. Front Mol Neurosci. April 2018; 11: 1–15, doi: 10.3389/fnmol.2018.00124.
- Kumari D, Swaroop M, Southall N, et al. “High-Throughput Screening to Identify Compounds That Increase Fragile X Mental Retardation Protein Expression in Neural Stem Cells Differentiated From Fragile X Syndrome Patient-Derived Induced Pluripotent Stem Cells,”. Stem Cells Transl Med. 2015; 4(7): 800–808, doi: 10.5966/sctm.2014-0278.
- Bodaleo F, Tapia-Monsalves C, Cea-Del Rio C, et al. “Structural and functional abnormalities in the olfactory system of fragile x syndrome models,”. Frontiers in Molecular Neuroscience. May 27, 2019; 12. Frontiers Media SA, doi: 10.3389/fnmol.2019.00135.
- van der Staay FJ, Arndt SS, Nordquist RE. “Evaluation of animal models of neurobehavioral disorders,”. Behav Brain Funct. 2009; 5: 1–23, doi: 10.1186/1744-9081-5-11.
- Lancaster MA, Renner M, Martin CA, et al. “Cerebral organoids model human brain development and microcephaly,”. Nature. 2013; 501(7467): 73–379, doi: 10.1038/nature12517.
- Chen HI, Song H, li Ming G. “Applications of Human Brain Organoids to Clinical Problems,”. Developmental Dynamics. Jan 01, 2019; 248(1): 53–64. John Wiley and Sons Inc, doi: 10.1002/dvdy.24662.
- Vershkov D, Fainstein N, Suissa S, et al. “FMR1 Reactivating Treatments in Fragile X iPSC-Derived Neural Progenitors In Vitro and In Vivo,”. Cell Rep. 2019; 26, doi: 10.1016/j.celrep.2019.02.026.